一种砜吡草唑及其中间体的制备方法与流程
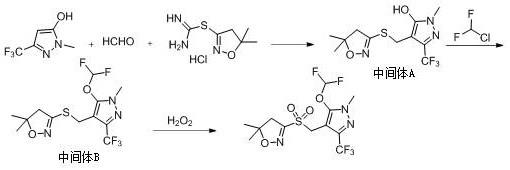
1.本发明涉及一种砜吡草唑中间体的制备方法,尤其涉及一种采用1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺和5,5-二甲基-3-巯基-4,5-二氢异恶唑合成砜吡草唑中间体a的方法,以及采用该新的工艺路线合成砜吡草唑的方法,属于砜吡草唑合成技术领域。
背景技术:
2.砜吡草唑(pyroxasulfone)是一种异噁唑类除草剂,由日本组合化学株式会社研究开发,化学名称为3-[5-(二氟甲氧基)-1-甲基-3-(三氟甲基)吡唑-4-基甲基磺酰基]-4,5-二氢-5,5-二甲基-1,2-异噁唑,分子式为c
12
h
14
f5n3o4s,分子量为391.32, cas登录号为447399-55-5。砜吡草唑可用于大多数作物田的芽前土壤处理剂,施用后被杂草幼根与幼芽吸收,抑制幼苗早期生长,破坏分生组织与胚芽鞘,是植物体内vlcfa(极长侧链脂肪酸)(c20~c30)生物合成中严重的潜在抑制剂。其结构如下:目前,砜吡草唑的合成工艺报道的不多,专利cn102666502中公开了以下工艺路线:在碱性条件下,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛水溶液和5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐在水中进行缩合反应得到中间体a,中间体a再与二氟一氯甲烷烷基化得到中间体b,中间体b经双氧水氧化,得到砜吡草唑。
[0003]
上述工艺路线中,砜吡草唑收率过低,且甲醛水溶剂易致畸,对环境和人类危害大。
技术实现要素:
[0004]
针对现有技术存在的不足,本发明提供了一种砜吡草唑中间体a的制备方法,该方法为中间体a的合成提供了一种新的思路。
[0005]
本发明提供了一种式a所示的砜吡草唑中间体a的制备方法,所述中间体a由1-甲
基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺和5,5-二甲基-3-巯基-4,5-二氢异恶唑在酸存在下反应得到;反应式如下:。
[0006]
进一步的,上述制备方法中,所述环仲胺为能够与1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛反应形成曼尼希碱的环仲胺,其具有饱和的或不饱和的五元环或六元环,仲胺基位于环上,环上带有除氮以外的杂原子或不带除氮以外的杂原子,环上带有取代基或不带取代基。优选的,环仲胺为吡咯、吗啉或哌啶。
[0007]
进一步的,上述制备方法中,反应在酸存在下进行。所述酸为能与环仲胺形成盐的无机酸,例如盐酸、硫酸等。酸与环仲胺按照等摩尔的量加入,或者酸可以稍微过量。
[0008]
进一步的,上述制备方法中,反应在溶剂存在下进行。各反应物在水和有机溶剂的环境中进行反应,所述水是由酸带入的,所述有机溶剂选择能够与水互溶保持反应均相进行且不与各反应物和产物进行反应的有机溶剂,例如醇类溶剂等,基于环保和成本等考虑,所用有机溶剂优选为乙醇。
[0009]
进一步的,上述制备方法中,各有机溶剂对反应的进行影响不大,有机溶剂的用量无特殊要求,能保证各反应物均相反应、反应较为容易的进行、反应容器的利用率较高即可。
[0010]
进一步的,本发明所用的1-甲基-3-(三氟甲基)-1h-吡唑-5-醇可以根据现有技术中报道的方法进行制备,例如按照文献:journal of agricultural and food chemistry (2008), 56(22), 10805-10810中公开的方法进行制备。5,5-二甲基-3-巯基-4,5-二氢异恶唑可以市购。
[0011]
进一步的,反应时,先将1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺在酸存在下进行曼尼希(mannich)反应,然后加入5,5-二甲基-3-巯基-4,5-二氢异恶唑进行反应,得到中间体a。1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺、酸、溶剂的加入顺序没有严格的要求,可以按照随意的加入顺序进行混合,可以一次性加入,也可以分批次的加入,也可以连续性的加入,可以以纯物质的形式加入,也可以先溶于溶剂中以溶液的形式加入。5,5-二甲基-3-巯基-4,5-二氢异恶唑可以一次性加入,也可以分批次的加入,也可以连续性的加入,可以以纯物质的形式加入,也可以先溶于溶剂中以溶液的形式加入。
[0012]
进一步的,上述制备方法中,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺和5,5-二甲基-3-巯基-4,5-二氢异恶唑可以按照理论摩尔比进行反应,优选在多聚甲醛、环仲胺和5,5-二甲基-3-巯基-4,5-二氢异恶唑稍过量的情况下进行反应。在本发明某一具体实施方式中,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺和5,5-二甲基-3-巯基-4,5-二氢异恶唑按照1:1.0-1.5:1.0-1.5:1.0-1.3的摩尔比进行反应,多聚甲醛的摩尔量以甲醛计。
[0013]
进一步的,上述制备方法中,曼尼希反应的温度为60℃~80℃,在此反应温度下可以使反应较快速的进行,反应时间因范围温度的不同稍有差异,当原料含量极低时判定反应结束。加入5,5-二甲基-3-巯基-4,5-二氢异恶唑后,也在60℃~80℃下进行反应。
[0014]
进一步的,上述制备方法中,先将1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺、酸、溶剂混合,在60℃~80℃进行反应,至1-甲基-3-(三氟甲基)-1h-吡唑-5-醇反应完全后再加入5,5-二甲基-3-巯基-4,5-二氢异恶唑继续保温反应,直至5,5-二甲基-3-巯基-4,5-二氢异恶唑反应完全。可以通过hplc法来检测反应的进度。
[0015]
进一步的,上述制备方法中,反应液的后处理简单、粗放,反应后将反应液去除有机溶剂,然后加入适量的水搅拌析晶,抽滤即可得到最终产物。反应液可以通过蒸馏、减压蒸馏、旋蒸等方式去除有机溶剂。
[0016]
本发明还提供了一种砜吡草唑的制备方法,该方法包括按照上述方法合成中间体a的步骤。
[0017]
进一步的,上述合成砜吡草唑的方法中,除了合成中间体a的步骤外,还包括中间体a与二氟一氯甲烷进行烷基化反应得到中间体b、以及中间体b与双氧水进行氧化反应得到砜吡草唑的步骤。整个反应过程的工艺路线如下:。
[0018]
进一步的,合成中间体b以及由中间体b合成砜吡草唑的具体工艺过程可以按照现有技术中报道的方法进行,例如可以按照专利cn102666502a中描述的方式进行,这一部分属于现有技术,在此不再赘述。
[0019]
在本发明某一具体实施例中,中间体a与二氟一氯甲烷的反应在有机溶剂和缚酸剂存在下进行,缚酸剂为现有技术中报道的任意能够将反应形成的酸消耗掉的物质,常用的为碳酸钠、碳酸钾、三乙胺。有机溶剂为现有技术中报道的可行的有机溶剂,例如乙腈等。
[0020]
在本发明某一具体实施例中,中间体a与二氟一氯甲烷的摩尔比为1:1.2-3,反应温度为10℃~30℃,反应过程中时时通过hplc检测中间体a的含量,当中间体a完全反应时反应结束。反应结束后,过滤去除副产物盐,反应液无须提取产品直接可以用于下一步反应。
[0021]
在本发明某一具体实施例中,中间体b与过氧化氢的摩尔比为1:5~10,中间体b与双氧水的反应温度为60℃-80℃。双氧水的浓度可以随意选择,一般选择较高浓度。反应过程中时时通过hplc检测中间体b含量,当中间体b完全反应时反应结束。反应结束后,减压蒸馏去除有机溶剂,然后过滤产生的固体,即为砜吡草唑。
[0022]
本发明的优点在于:(1)以1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、多聚甲醛、环仲胺和5,5-二甲基-3-巯基-4,5-二氢异恶唑反应得到中间体a,为中间体a的合成提供了新的思路。
[0023]
(2)中间体a合成过程中以多聚甲醛代替易致畸的甲醛水溶剂,更为绿色环保。
[0024]
(3)中间体a合成的反应条件温和,不需要过高的温度和高压,对设备要求低。
[0025]
(4)中间体a的后处理简单粗放,反应产物容易分离、简化了生产工艺,更适合工业化大生产。
[0026]
(5)中间体a的反应无须特殊的催化剂,反应选择性高,反应收率较高,产物纯度较高,砜吡草唑的总收率在81%以上。
[0027]
具体实例方式以下结合具体实施例对本发明做进一步说明。下述说明仅是示例性的,并不对保护范围进行限制。在不脱离本发明发明构思的基础上,本领域技术人员不经创造性劳动所得的其他实施方案,也在保护范围之内。
[0028]
下述实施例中,如无特别说明,所述浓度均为质量百分浓度。
[0029]
下述实施例中,所用原料如无特别说明,均为市购产品。
[0030]
下述实施例中,收率=产品的实际质量
×
纯度/产品的理论质量。
[0031]
实施例11、中间体a合成:室温下在40ml乙醇中依次加入1-甲基-3-(三氟甲基)-1h-吡唑-5-醇16.6g、多聚甲醛3g、哌啶8.5g、37%盐酸水溶液10g,加热至80℃搅拌反应4hr,hplc检测无1-甲基-3-(三氟甲基)-1h-吡唑-5-醇剩余后加入5,5-二甲基-3-巯基-4,5-二氢异恶唑21.6g,继续反应,继续反应4hr后hplc检测5,5-二甲基-3-巯基-4,5-二氢异恶唑反应完全,反应结束后减压蒸除乙醇,加入20ml水打浆2hr,抽滤得白色固体沉淀,干燥得28.8g固体,即为中间体a,hplc测得纯度为99.4%,收率以1-甲基-3-(三氟甲基)-1h-吡唑-5-醇计为92.6%。
[0032]
2、中间体b合成:向40ml乙腈中加入6.9g碳酸钾和14.4g中间体a,20℃搅拌2hr,常压下缓缓通入二氟一氯甲烷气体8.5g,8hr后hplc检测中间体a反应完全,反应结束后过滤除去固体,母液直接用来下一步反应。
[0033]
3、砜吡草唑合成:室温下往上述步骤2的反应液中加入30wt%双氧水28.3g,搅拌1hr后,缓缓升温至60℃进行反应,4hr后hplc检测显示反应结束。减压浓缩除去乙腈后,反应液冷却至室温,过滤得白色固体16.8g,即为砜吡草唑,经hplc检测其含量为98.4%以中间体a计,砜吡草唑收率为84.5%。
[0034]
实施例21、中间体a合成:室温下在80ml乙醇中依次加入1-甲基-3-(三氟甲基)-1h-吡唑-5-醇33.2g、多聚甲醛6g、吡咯14.2g、37%盐酸水溶液20g,加热至75℃搅拌反应4hr,hplc检测无1-甲基-3-(三氟甲基)-1h-吡唑-5-醇剩余后加入5,5-二甲基-3-巯基-4,5-二氢异恶唑43.2g,继续反应,继续反应4hr后hplc检测反应结束,将反应液减压蒸除乙醇,加入20ml水打浆2hr,抽滤得白色固体沉淀,干燥得57.5g固体,即为中间体a,hplc测得纯度为99.2%,收率以1-甲基-3-(三氟甲基)-1h-吡唑-5-醇计为92.3%。
[0035]
2、中间体b合成:向40ml乙腈中加入10.6g碳酸钠和30.9g 中间体a,20℃搅拌2hr,常压下缓缓通入二氟一氯甲烷气体15g,8hr后hplc检测中间体a完全反应,反应结束后过滤除去固体,母液直接用来下一步反应。
[0036]
3、砜吡草唑合成:
室温下往上述步骤2的反应液中加入30wt%的双氧水113.3g,搅拌1hr后,缓缓升温至60℃进行反应,4hr后hplc检测显示反应结束。减压浓缩除去乙腈后,冷却至室温,过滤得白色固体34.1g,即为砜吡草唑,经hplc检测其含量为98.1%,以中间体a计,砜吡草唑收率为87.5%。
[0037]
实施例3中间体a合成:室温下在80ml乙醇中依次加入1-甲基-3-(三氟甲基)-1h-吡唑-5-醇33.2g、多聚甲醛6g、吗啉17.4g、37%盐酸水溶液20g,加热至60℃搅拌反应4hr,hplc检测无1-甲基-3-(三氟甲基)-1h-吡唑-5-醇剩余后加入5,5-二甲基-3-巯基-4,5-二氢异恶唑43.2g,继续反应,继续反应4hr后hplc检测反应结束,反应后减压蒸除乙醇,加入20ml水打浆2hr,抽滤得白色固体沉淀,干燥得56.8g固体,即为中间体a, hplc测得纯度为99.3%,收率以1-甲基-3-(三氟甲基)-1h-吡唑-5-醇计为91.3%。
[0038]
2、中间体b合成:向40ml乙腈中加入10.6g碳酸钠和30.9g 中间体a,20℃搅拌2hr,常压下缓缓通入二氟一氯甲烷气体16g,8hr后hplc检测中间体a反应完全,反应结束后过滤除去固体,母液直接用来下一步反应。
[0039]
3、砜吡草唑合成:室温下往上述步骤2的反应液中加入30wt%的双氧水56.6g,搅拌1hr后,缓缓升温至80℃进行反应,4hr后hplc检测显示反应结束。减压浓缩除去乙腈后,冷却至室温,过滤得白色固体33.1g,即为砜吡草唑,经hplc检测其含量为98.7%,以中间体a计,砜吡草唑收率为83.5%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1