一种丙硫菌唑中间体的绿色合成方法与流程
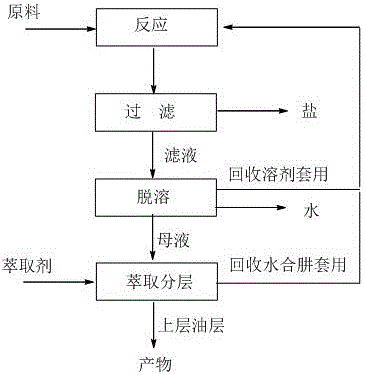
本发明涉及一种丙硫菌唑中间体的合成方法,具体涉及一种丙硫菌唑中间体的绿色合成方法。
背景技术:
丙硫菌唑是拜耳公司2004年开发上市的一种新型三唑硫酮类杀菌剂,毒性低,无致畸、致突变性,对胚胎无毒性,对人和环境安全。其作用机理是抑制真菌中甾醇的前体——羊毛甾醇或2,4-亚甲基二氢羊毛甾14位上的脱甲基化作用,不仅具有很好的内吸活性,优异的保护、治疗和根除活性,且持效期长,具有广谱的杀菌活性。
2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼是合成丙硫菌唑的关键中间体,其由2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇和水合肼反应得到,在专利us6201128、us6559317、cn109553584a中均匀涉及。但是这些现有技术中合成2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼的反应有诸多不足,具体如下:
us6201128中使用10倍当量的水合肼,在稀释剂存在下与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇进行反应。水合肼同时作为反应物和碱及稀释剂,反应后产生大量低浓度水合肼废液,增加了生产成本。后处理过程中需要水洗、干燥,操作步骤繁琐。
us6559317也使用了大大过量的水合肼进行反应。反应结束后通入氯化氢,得到2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼的盐酸盐,在后续的反应中再加入碱进行游离。该法产生了大量的低浓度水合肼废水,此外使用成盐-游离的方式带来了含盐废水的处理问题,后处理步骤繁琐,不利于工业化生产的进行。
cn109553584a将2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇和水合肼在100℃下搅拌反应,至完全转化后停止反应,反应液冷却至室温,用饱和食盐水洗涤,甲苯萃取3次,合并有机相,向有机相中加入盐酸,搅拌后静置析晶,过滤得到2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼的盐酸盐,收率73%。此方法使用饱和食盐水洗涤水合肼,产生大量的低浓度水合肼废水,回收难度大,生产成本高。
目前,现有的合成方法中存在大量水合肼废水的产生、废水处理操作步骤复杂的缺陷,因此开发一种该中间体的绿色合成工艺对于丙硫菌唑的工业化生产具有非常重要的意义和价值。
技术实现要素:
本发明的目的在于提供一种丙硫菌唑中间体——2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼的绿色合成方法,该方法原料转化率高,产物选择性高,过量的水合肼可以循环套用,大大简化了整个工艺流程,符合绿色环保的要求。
为达此目的,本发明采用以下技术方案:
一种丙硫菌唑中间体的绿色合成方法,该方法包括以下步骤:
(1)2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇、水合肼、相转移催化剂和缚酸剂在溶剂中进行反应;
(2)反应后,将反应液过滤,先通过脱除溶剂的方式浓缩反应液,然后对浓缩后的反应液进行萃取,得到上层丙硫菌唑中间体相和下层水合肼相,所得水合肼相作为水合肼原料回用。
本发明反应式如下:
上述方法中,反应后处理简单,反应形成的盐通过过滤的方式可以直接去除,脱溶得到的溶剂可以循环利用,萃取后的丙硫菌唑中间体相可以直接进入制备丙硫菌唑的下一步反应,萃取后的水合肼相中水合肼浓度高,可以直接套用于下一批反应中,作为水合肼原料使用。整个后处理过程简单、高效,无废水产生,绿色环保。
进一步的,本发明采用相转移催化剂提高原料的转化率,采用缚酸剂促进反应向正向进行,整个反应选择性高,产品的收率高。其中,所述相转移催化剂和缚酸剂在现有技术中有诸多报道,可以从现有技术中选择有此作用的物质用于本发明。例如,所述相转移催化剂可以为环状冠醚类催化剂或季铵盐类催化剂,优选为18冠6、四丁基溴化铵、四丁基氯化铵等。所述敷酸剂为可以与产生的酸反应形成盐的物质,例如碱金属氢氧化物、碱土金属氢氧化物、碱金属碳酸盐、碱土金属碳酸盐等,优选形成的盐能够通过过滤的方式从反应液中去除的敷酸剂,例如氢氧化钾、氢氧化钠、氢氧化钙、碳酸钾、碳酸钠、碳酸钙等。
进一步的,步骤(1)中,敷酸剂与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇的摩尔比为(0.5~2):1,优选为(1~1.5):1。
进一步的,步骤(1)中,相转移催化剂与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇的质量比为(0.1%-2%):1,优选为(0.5%-1%):1。
进一步的,步骤(1)中,水合肼可以是纯的水合肼,也可以是水合肼水溶液。工业上的水合肼原料大多是水合肼水溶液。为了降低副反应的发生、提高反应收率、同时保证反应的安全性,水合肼优选以水溶液的形式加入。水合肼水溶液的浓度优选大于等于60wt%,例如60-100wt%(不包括100%),当浓度为100wt%时,表示是纯的水合肼。优选的,水合肼水溶液的浓度为70-80wt%。
进一步的,步骤(1)中,水合肼与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇的摩尔比为(1~10):1,优选为(5~8):1。
进一步的,步骤(1)中,优选先将2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇、水合肼、相转移催化剂溶于溶剂中,然后加入敷酸剂进行反应。缚酸剂缓慢的加入体系中,一般加入时间为2-3h。加入敷酸剂的过程中以及加入后,控制反应温度为40-100℃进行反应,优选控制温度为60-90℃。反应进行可以通过时时检测原料的消耗量来评判,原料转化完全后结束反应。
进一步的,本发明的溶剂为水和有机溶剂,其中水是通过水合肼引入体系中的,水的含量太多不利于反应的收率提高,通过控制水合肼的浓度来控制体系中水的用量,水合肼的浓度不低于60wt%。所述有机溶剂常用的有c1-c3醇类溶剂,例如甲醇、乙醇、丙醇中的任意一种或多种。
优选的,有机溶剂与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇的质量比为(1~8):1,优选为(3~5):1。
进一步的,本发明水合肼优选过量加入,反应过程中消耗了部分水合肼,同时有水产生,导致反应结束后水合肼浓度降低。浓度过低的水合肼无法回用于生产,否则会大大降低反应产率,增加副反应的发生。本发明在后处理过程中,先通过过滤除去反应形成的盐,然后对反应液进行脱溶,随着溶剂的脱出,反应液也被浓缩,起到了浓缩水合肼的作用,通过控制脱溶的程度可以使水合肼的浓度在反应前后不发生明显变化,然后对浓缩后的反应液进行萃取,萃取得到的丙硫菌唑中间体相可以直接进入制备丙硫菌唑的下一步反应,萃取得到的水合肼相可以直接套用,脱溶所得的溶剂也可以正常回收套用。
进一步的,脱溶的过程中,可以通过常压蒸馏、减压蒸馏、旋蒸等现有技术常用的手段进行,脱溶至反应液中水合肼浓度大于等于60wt%时停止,然后进行萃取,优选脱溶至反应液中水合肼浓度大于等于70-80wt%时停止。脱溶的过程中,先脱除有机溶剂,然后再脱除水,直至水合肼浓度符合要求。
进一步的,步骤(2)中,所述萃取剂为芳香烃、酮类溶剂,优选为甲苯、二甲苯、甲基异丁基酮、甲基异丙基酮中的任意一种或多种。
优选地,所述萃取剂与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇的质量比为(1~8):1,优选为(2~4):1。
本发明具有以下有益效果:
(1)本发明采用相转移催化剂和敷酸剂,可使原料2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇彻底转化,减小了副反应选择性,提高了产物2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼的选择性,使反应收率大大提高。
(2)本发明条件温和,工艺操作简单,后处理方便、高效,水合肼层可直接套用于下一批反应,脱溶后得到的溶剂也可以正常回收套用,是一条绿色合成工艺。
(3)萃取后得到的2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼溶液不需要纯化分离,可直接进入下一步反应,简化了操作流程,有利于工业化生产。
附图说明
图1本发明丙硫菌唑中间体的合成流程图。
具体实施方式
下面通过具体的实施例对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
本发明实施例中对丙硫菌唑中间体2-(1-氯-环丙烷)-3-(2-氯苯基)-2-羟基]-丙烷-1-肼的含量测定均采用高效液相色谱法。
下述实施例中,如无特别说明,水合肼的浓度均为质量百分浓度,2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇的纯度为95%。
实施例1
在配有冷凝器的烧瓶中,依次投入乙醇300g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、80%水合肼212.6g(3.4mol)、四丁基氯化铵0.5g,开启搅拌,控温在80-85℃,开始加入碳酸钾48g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,分别得到前馏分乙醇295g,后馏分水10.4g。剩余物中加入甲苯200g,搅拌30min后静置分层,得到上层油层299.0g,取样经hplc测定产物含量30.46%,收率97.4%。下层水合肼层191.8g(3.06mol),分析测定水合肼含量79.8%,可直接套用于下一批反应。
实施例2
在配有冷凝器的烧瓶中,依次投入新鲜乙醇15g和前述的回收乙醇285g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、20.8g新鲜80%水合肼和191.8g(3.06mol)前述回收的水合肼、四丁基氯化铵0.5g,开启搅拌,控温在80-85℃,开始加入碳酸钾48g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,分别得到前馏分乙醇293g,后馏分水10.4g。剩余物中加入甲苯200g,搅拌30min后静置分层,得到上层油层298.9g,取样经hplc测定产物含量30.53%,收率97.6%。下层水合肼层191.8g,分析测定水合肼含量79.8%,可直接套用于下一批反应。
实施例3
在配有冷凝器的烧瓶中,依次投入甲醇300g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、80%水合肼106.3g(1.7mol)、四丁基溴化铵0.5g,开启搅拌,控温在60-65℃,开始加入氢氧化钾19.3g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,将溶剂脱除,分别得到前馏分甲醇297g,后馏分水10.8g。剩余物中加入二甲苯200g,搅拌30min后静置分层,得到上层油层298.7g,取样经hplc测定产物含量30.00%,收率95.8%。下层水合肼层91.2g(1.36mol),分析测定水合肼含量74.6%,可直接套用于下一批反应。
实施例4
在配有冷凝器的烧瓶中,依次投入甲醇300g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、80%水合肼106.3g(1.7mol)、四丁基溴化铵0.3g,开启搅拌,控温在40-45℃,开始加入氢氧化钾19.3g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,将溶剂脱除,分别得到前馏分甲醇295g,后馏分水16.5g。剩余物中加入二甲苯200g,搅拌30min后静置分层,得到上层油层298.6g,取样经hplc测定产物含量29.43%,收率94.0%。下层水合肼油层85.5g,分析测定水合肼含量79.7%,可直接套用于下一批反应。
实施例5
在配有冷凝器的烧瓶中,依次投入丙醇300g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、70%水合肼194.4g(2.72mol)、0.5g18冠6,开启搅拌,控温在95-100℃,开始加入氢氧化钠13.7g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,将溶剂脱除,分别得到前馏分丙醇295g,后馏分水19.5g。剩余物中加入甲基异丁基酮200g,搅拌30min后静置分层,得到上层油层298.7g,取样经hplc测定产物含量30.11%,收率96.2%。下层水合肼层170.6g(2.38mol),分析测定水合肼含量69.8%,可直接套用于下一批反应。
实施例6
在配有冷凝器的烧瓶中,依次投入丙醇500g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、226.7g(2.38mol)60%水合肼、0.5g18冠6,开启搅拌,控温在85-90℃,开始加入氢氧化钠13.7g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,将溶剂脱除,分别得到前馏分丙醇491g,后馏分水23.6g。剩余物中加入甲基异丙基酮200g,搅拌30min后静置分层,得到上层油层298.7g,取样经hplc测定产物含量28.80%,收率92.0%。下层水合肼层199.2g,分析测定水合肼含量59.9%,可直接套用于下一批反应。
实施例7
在配有冷凝器的烧瓶中,依次投入乙醇500g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、80%水合肼106.3g(1.7mol)、四丁基溴化铵0.5g,开启搅拌,控温在80-85℃,开始加入氢氧化钠13.7g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,分别得到前馏分乙醇490g,后馏分水16.5g。剩余物中加入甲苯200g,搅拌30min后静置分层,得到上层油层298.6g,取样经hplc测定产物含量30.90%,收率98.7%。下层水合肼层85.5g(1.36mol),分析测定水合肼含量79.5%,可直接套用于下一批反应。
实施例8
在配有冷凝器的烧瓶中,依次投入乙醇400g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、80%水合肼106.3g(1.7mol)、四丁基溴化铵1.0g,开启搅拌,控温在80-85℃,开始加入氢氧化钾19.3g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,将溶剂脱除脱除,分别得到前馏分乙醇387g,后馏分水8.3g。剩余物加入甲苯200g,搅拌30min后静置分层,得到上层油层298.7g,取样经hplc测定产物含量30.68%,收率98%。下层水合肼层93.7g(1.36mol),分析测定水合肼含量72.2%,可直接套用于下一批反应。
实施例9
在配有冷凝器的烧瓶中,依次投入甲醇400g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、80%水合肼170.1g(2.72mol)、四丁基氯化铵0.5g,开启搅拌,控温在60-65℃,开始加入氢氧化钾19.3g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,分别得到前馏分甲醇392g,后馏分水33g。剩余物加入甲苯200g,搅拌30min后静置分层,得到上层油层298.7g,取样经hplc测定产物含量30.55%,收率97.6%。下层水合肼层132.8g(2.38mol),分析测定水合肼含量89.7%,可直接套用于下一批反应。
对比例1
在配有冷凝器的烧瓶中,依次投入甲醇500g、2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇100g(0.34mol)、50%水合肼272g(2.72mol)、四丁基氯化铵0.5g,开启搅拌,控温在60-65℃,开始加入氢氧化钾19.3g,加料时间2h,加料完毕后,继续保温反应至原料转化完全。反应完毕后,降温至室温,反应液过滤除盐,进行减压脱溶,分别得到前馏分甲醇393g,后馏分水29.3g。剩余物加入甲苯200g,搅拌30min后静置分层,得到上层油层270.1g,取样经hplc测定产物含量23.47%,收率75%。下层水合肼油层238.6g(2.38mol),分析测定水合肼含量49.9%。
通过实施例9与对比例1的对比可知,水合肼的浓度降低至50%,产物收率降低明显。
如上所述,尽管参照特定的优选实施例已经表示和表述了本发明,但并不得解释对本发明自身的限制。在不脱离所附权利要求定义的本发明的精神和范围前提下,可对其在形式上和细节上作出各种变化。
- 还没有人留言评论。精彩留言会获得点赞!