一种含苯基化合物的制备方法与流程
1.本发明涉及医药技术领域,尤其涉及一种含苯基化合物的制备方法。
背景技术:
2.替格瑞洛(ticagrelor),中文化学名((1s,2s,3r,5s)-3-[7-[[(1r,2s)-2-(3,4-二氟苯基)环丙基]氨基]-5-丙硫基三唑并[4,5-d]嘧啶-3-基]-5-(2-羟乙氧基)-[0003]
1,2-环戊二醇)。阿斯利康公司研发的一种口服血小板聚集抑制剂药物。该药可逆的作用于adp受体p2y12,对adp引起的血小板聚集有较好的抑制作用,口服起效迅速,临床上用于急性冠脉综合征(不稳定性心绞痛/非st抬高性心肌梗死/st抬高性心肌梗死),降低血栓性心血管事件的发生率。替格瑞洛结构式如下:
[0004][0005]
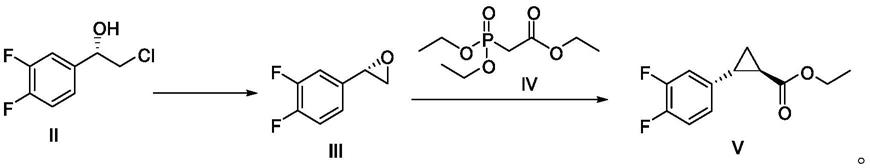
现有技术中通过化合物(ⅱi)与膦酰基乙酸三乙酯经环丙烷化反应制得化合物(
ⅴ
),化合物(
ⅴ
)经胺酯交换,霍夫曼降解制得化合物(
ⅵ
),然而在将化合物(v)胺酯交换反应中,目前报道的胺酯交换的合成方法主要有如下几种;
[0006][0007]
1)cn 104326922a报道了直接采用氨甲醇溶液进行胺酯交换反应,该方法主要缺点是反应速率较慢t=60℃反应18h,原料剩余约80%。
[0008]
2)cn 106906249a报道了氨甲醇溶液中加入甲醇钠加快反应,但是实验发现该方法导致酰胺水解杂质较大,不适合工业化生产。
[0009]
3)wo 2008018822a1报道了氨甲醇溶液、甲醇钠、甲酸甲酯进行胺酯交换反应,该方法反应结果良好,但对反应设备有耐压要求,且存在安全隐患。
[0010]
因此本领域需要一种更有利于工业化生产且操作简单的,并且高产率制得化合物
(ⅰ)的方法,从而快速有效的制备得到替格瑞洛关键中间体(1r,2s)-2-(3,4-二氟苯基)环丙胺。
技术实现要素:
[0011]
本发明要解决的技术问题是为了克服现有的含苯基化合物制备方法工艺复杂、反应时间长,条件苛刻且产率低的问题,本发明提供了与现有技术不同的一种含苯基化合物的制备方法,具体的为替格瑞洛中间体的制备方法。该方法工艺简单,反应时间短、易操作、成本低、适合工业化生产,且产物产率高。
[0012]
本发明提供了一种如式i所示的化合物的制备方法,其包括如下步骤:
[0013]
在甲酰胺或乙酰胺存在下中,在碱存在下,如式v所示的化合物进行如下所述的胺酯交换反应,得如式i所示的化合物,即可;
[0014][0015]
所述的碱为甲醇钠、乙醇钠或甲醇锂。
[0016]
在所述的胺酯交换反应中,所述的碱优选甲醇钠。
[0017]
在所述的胺酯交换反应中,所述的如式v所示的化合物与所述的碱的摩尔比可为本领域该类胺酯交换反应常规的比例,优选1:1~1:5,更优选1:1.4。
[0018]
在所述的胺酯交换反应中,所述的如式v所示的化合物在甲酰胺或乙酰胺中的浓度可为1mol/l~5mol/l(例如2.75mol/l)。
[0019]
在所述的胺酯交换反应中,所述的化合物v的纯度可为90%~99.9%,例如95%。
[0020]
在所述的胺酯交换反应中,所述的反应温度可为本领域该类胺酯交换反应常规的温度,优选0℃~100℃,更优选60℃。
[0021]
在所述的胺酯交换反应中,所述的反应进程可采用本领域常规的方法进行监测(例如tlc,hplc),以如式v所示的化合物消失为反应终点,所述的反应时间优选1h~10h,更优选4h~5h。
[0022]
所述的胺酯交换反应结束后,还可包含后处理,所述的后处理优选包含如下步骤:加水,冷却,过滤,干燥,得所述的如式v所示的化合物,即可。
[0023]
在某一实施方案中,所述的胺酯交换反应的原料仅包含所述的甲酰胺或乙酰胺、所述的碱和所述的如式v所示的化合物。
[0024]
在某一实施方案中,所述的如式i所述的化合物的制备方法可为如下任一方案:
[0025]
方案一:
[0026]
所述的碱为甲醇钠,所述的如式v所示的化合物与所述的碱的摩尔比为1:4;
[0027]
方案二:
[0028]
所述的碱为甲醇钠,所述的反应温度为60℃,所述的如式v所示的化合物在甲酰胺或乙酰胺中的浓度为2.75mol/l;
[0029]
方案三:
[0030]
所述的碱为甲醇钠,所述的如式v所示的化合物与所述的碱的摩尔比为1:4,所述
的反应温度为60℃;
[0031]
方案四:
[0032]
所述的碱为甲醇钠,所述的如式v所示的化合物与所述的碱的摩尔比为1:4,所述的反应温度为60℃,所述的如式v所示的化合物在甲酰胺或乙酰胺中的浓度为2.75mol/l。
[0033]
所述的如式i所示的化合物的制备方法还可进一步包含如下步骤:
[0034]
步骤(1)在溶剂中,在碱存在下,如式ii所示的化合物进行如下所示的环合反应,得如式iii所示的化合物,即可;
[0035]
步骤(2)在溶剂中,在碱存在下,如式iii所示的化合物和如式iv所述的化合物进行如下所示的环丙烷化反应,得如式v所示的化合物,即可;
[0036][0037]
在所述的步骤(1)中,所述的溶剂可为本领域该类环合反应常规的溶剂,优选芳烃类溶剂、或、芳烃类溶剂和水,更优选芳烃类溶剂和水;所述的芳烃类溶剂优选甲苯。
[0038]
在所述的步骤(1)中,所述的碱可为本领域该类环合反应常规的碱,优选碱金属的氢氧化物,更优选氢氧化钠、氢氧化钾和氢氧化锂,最优选氢氧化钠。
[0039]
在所述的步骤(1)中,所述的如式ii所示的化合物与所述的碱的摩尔比可为本领域该类环合反应常规的摩尔比,优选1:1.0-1:3.0;更优选1:1.5。
[0040]
在所述的步骤(1)中,所述的如式ii所示的化合物在溶剂中的浓度可为本领域该类环合反应常规的浓度,优选1mol/l~1.5mol/l。
[0041]
在所述的步骤(1)中,所述的反应温度可为本领域该类环合反应常规的温度,优选0℃~50℃(例如10℃、20℃、25℃或30℃),更优选30℃。
[0042]
在所述的步骤(1)中,所述的环合反应的进程可采用本领域常规的方法进行监测(例如tlc,hplc),以如式ii所示的化合物消失为反应终点,所述的反应时间优选2h~10h,更优选5h~6h。
[0043]
在某一实施方案中,所述的步骤(1)中的环合反应的原料仅包含所述的溶剂、所述的碱和所述的如式ii所示的化合物。
[0044]
所述的步骤(1)还可包含后处理,所述的后处理优选包含如下步骤:在所述的环合反应结束后,萃取,干燥后直接进行所述的步骤(2)的反应。
[0045]
在所述的步骤(2)中,所述的环丙烷化反应可在惰性气体保护下进行,所述的惰性气体优选氮气。
[0046]
在所述的步骤(2)中,所述的溶剂可为本领域该类环丙烷化反应常规的溶剂,优选芳烃类溶剂,更优选甲苯。
[0047]
在所述的步骤(2)中,所述的碱可为本领域该类环丙烷化反应常规的碱,优选叔丁醇钾、叔丁醇钠和叔丁醇锂,更优选叔丁醇钾。
[0048]
在所述的步骤(2)中,所述的如式iii所述的化合物与所述的碱的摩尔比可为本领域该类环丙烷化反应常规的比例,优选1:1~1:2。
[0049]
在所述的步骤(2)中,所述的如式iii所述的化合物与所述的如式iv所述的化合物的摩尔比可为本领域该类环丙烷化反应常规的比例,优选1:1~1:4,更优选1:1~1:2。
[0050]
在所述的步骤(2)中,所述的反应温度可为本领域该类环丙烷化反应常规的温度,优选20℃~100℃,优选60℃~80℃。
[0051]
在所述的步骤(2)中,所述的如式iii所述的化合物在所述的溶剂中的浓度可为本领域该类环丙烷化反应常规的浓度,优选1mol/l~5mol/l(例如3.7mol/l)。
[0052]
在所述的步骤(2)中,所述的环丙烷化反应的反应进程可采用本领域常规的方法进行监测(例如tlc,hplc),以如式iii所示的化合物消失为反应终点,所述的反应时间优选8h~48h,更优选24h~48h。
[0053]
在某一实施方案中,所述的步骤(2)中的环丙烷化反应的原料仅包含所述的溶剂、所述的碱、所述的如式iii所示的化合物和所述的如式iv所述的化合物。
[0054]
所述的步骤(2)还可包含后处理,所述的后处理优选包含如下步骤:在所述的环丙烷化反应结束后,淬灭,加水,萃取,浓缩,得所述的如式v所示的化合物,即可。
[0055]
本发明还提供了一种如式vi所示的化合物的制备方法,其包括如下步骤:步骤
①
:如式i所示的化合物的制备方法如上所述;
[0056]
步骤
②
:在水中,在碱存在下,在次氯酸钠存在下,如式i所示的化合物进行如下所示的霍夫曼降解反应得如式vi所示的化合物,即可;
[0057][0058]
在所述的步骤
②
中,所述的碱可为本领域该类霍夫曼降解反应常规的碱,优选碱金属的氢氧化物,更优选为氢氧化钠。
[0059]
在所述的步骤
②
中,所述的如式i所示的化合物与所述的碱的摩尔比可为本领域该类霍夫曼降解反应常规的比例,优选1:5~1:8,例如1:6。
[0060]
在所述的步骤
②
中,所述的如式i所示的化合物与所述的次氯酸钠的摩尔比可为本领域该类霍夫曼降解反应常规的比例,优选1:1~1:4。
[0061]
在所述的步骤
②
中,所述的如式i所示的化合物在水中的浓度可为本领域该类霍夫曼降解反应常规的浓度,优选0.8mol/l~1.2mol/l,例如0.9mol/l。
[0062]
在所述的步骤
②
中,所述的反应温度可为本领域该类霍夫曼降解反应中常规的温度,优选0℃~80℃,优选20℃~60℃,例如40℃。
[0063]
在所述的步骤
②
中,所述的霍夫曼降解反应的反应进程可采用本领域常规的方法进行监测(例如tlc,hplc),以如式i所示的化合物消失为反应终点,所述的反应时间优选3h~6h。
[0064]
在一个实施方案中,所述的霍夫曼降解反应的原料仅包含所述的水,所述的碱,所述的次氯酸钠和所述的如式i所示的化合物。
[0065]
所述的步骤
②
还可包含后处理,所述的后处理优选包含如下步骤:所述的霍夫曼降解反应结束后,调节ph至7-9,萃取,浓缩得所述的如式vi所示的化合物,即可;优选使用酯类溶剂进行萃取,更优选使用乙酸乙酯;所述的萃取结束后优选经过饱和氯化钠水溶液
洗涤并过滤后再进行浓缩步骤。
[0066]
本发明还提供了一种如式vi所示的化合物的盐酸盐的制备方法,其包括如下步骤:
[0067]
步骤a:如式vi所示的化合物的制备方法如上所述;
[0068]
步骤b:将如式vi所示的化合物与hcl进行成盐反应,得如式vi所示的化合物的盐酸盐,即可。
[0069]
所述的步骤a中还可包含后处理,所述的后处理优选包含如下步骤:所述的霍夫曼降解反应结束后,调节ph至7-9,萃取,萃取后进行所述的步骤b,即可;优选使用酯类溶剂进行萃取,更优选使用乙酸乙酯;所述的萃取结束后优选经过饱和氯化钠水溶液洗涤并过滤后再进行所述的步骤b。
[0070]
在所述的步骤b中,所述的如式vi所示的化合物与所述的hcl的比例可为1:1。
[0071]
在所述的步骤b中,所述的成盐反应可在酯类溶剂存在下进行,所述的酯类溶剂优选乙酸乙酯。
[0072]
本发明还提供了一种如式vi所示的化合物的d-扁桃酸盐的制备方法,其包括如下步骤:
[0073]
步骤i:如式vi所示的化合物的制备方法如上所述;
[0074]
步骤ii:在异丙醇中,将如式vi所示的化合物与d-扁桃酸进行成盐反应,得如式vi所示的化合物的d-扁桃酸盐,即可。
[0075]
所述的步骤i中还可包含后处理,所述的后处理优选包含如下步骤:所述的霍夫曼降解反应结束后,调节ph至7-9,萃取,浓缩后进行所述的步骤ii即可。
[0076]
在所述的步骤ii中,所述的如式vi所示的化合物与所述的d-扁桃酸的摩尔比可为1:1。
[0077]
在所述的步骤ii中,所述的成盐反应的温度可为本领域该类成盐反应的常规温度,例如45℃。
[0078]
在所述的步骤ii中,所述的d-扁桃酸盐在所述的异丙醇中的浓度可为本领域该类成盐反应的常规浓度,优选1mol/l~2mol/l,例如1.2mol/l。
[0079]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0080]
本发明所用试剂和原料均市售可得。
[0081]
本发明的积极进步效果在于:本发明提供了一种新的含苯基化合物的制备方法。该方法工艺简单,反应时间短、易操作、成本低、适合工业化生产,且产物产率高。
附图说明
[0082]
图1为对比例1中的反应结束后反应液的hplc图谱
具体实施方式
[0083]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。本发明的具体实施方式中,未作特别说明的技术手段或方法等为本领域的
常规技术手段或方法等。未作特别说明,本发明所用的原料和试剂均为市售产品。除非另外说明,否则所有的百分数、比率、比例、或份数按重量计。
[0084]
实施例1(2s)-2-(3,4-二氟苯基)环氧乙烷的制备(ⅲ)
[0085][0086]
1l三口烧瓶中加入化合物ⅱ(100g,0.52mol),甲苯(500ml)。机械搅拌,于10-30℃滴加10%氢氧化钠溶液312.5g,约1h滴加完毕。t=20-25℃保温反应5h,hplc监控原料<0.5%即可后处理。静置分层,水层用甲苯(200ml)萃取一次,合并有机层,饱和氯化钠水溶液洗涤一次。分出有机层,无水硫酸钠干燥化合物ⅲ甲苯溶液,不经纯化直接投入下一步反应,hplc纯度97.2%。
[0087]
实施例2(1r,2r)-2-(3,4-二氟苯基)环丙基甲酸乙酯(
ⅴ
)的制备
[0088][0089]
氮气保护下,上述制得的化合物ⅲ甲苯溶液(约700ml)、叔丁醇钠(60.1g,0.62mol,1.2eq)加入到干燥的1l三口烧瓶中搅拌(混悬液)。控制温度在20-30℃,缓慢滴加化合物ⅳ(140.1g,0.62mol,1.2eq)。滴加完毕后,t=20-30℃搅拌10min,然后升温至t=60℃反应48h。hplc监控化合物ⅲ<0.5%即可淬灭反应。将反应液冷却至t=5-10℃,滴加水(300ml)。静置分层,分出上层有机层,下层水层再次用甲苯(150ml)萃取一次,合并有机层。有机层无水硫酸钠干燥,浓缩至干得棕色油状物123.6g,油状物中产物含量93%,产物不经纯化直接进行下一步反应,hplc纯度95.3%,收率104.9%(以化合物ii计)(粗品推测其他杂质和溶剂残留导致实际质量收率高于理论收率,含量折算后该步反应实际摩尔收率95%)。
[0090]
实施例3(1r,2r)-2-(3,4-二氟苯基)环丙基甲酰胺(ⅰ)的制备
[0091][0092]
1l三口烧瓶中依次加入甲酰胺200ml,甲醇钠42.2g,化合物
ⅴ
(粗品123.6g)。机械搅拌,升温至t=60℃反应5h。原料反应完全后将搅拌下滴加水600ml,析出类白色固体。滴加完毕后缓慢降温冷却至t=0-5℃析晶3h。过滤,滤饼用纯化水(200ml)洗涤。固体于50℃鼓风干燥箱中干燥至恒重,得类白色固体化合物ⅰ(88.4g,化学纯度98.5%,收率86.2%,收率以化合物ⅱ计)。1h nmr(dmso-d6)δ:1.16~1.23(m,1h),1.29~1.34(m,1h),1.79~1.85(m,1h),2.24~2.26(m,1h),5.73(s,2h),6.79~6.87(m,2h),6.89~7.01(m,1h);
13
c nmr
(101mhz,dmso-d6)δ15.81,24.20,25.22,114.55,116.80,121.82,137.10,147.40,148.52,149.72,150.99,173.42.
[0093]
hplc条件:色谱柱:gemini c18 110r(250mm*4.6mm,5um);流动相:a:0.1%三氟乙酸-水,b:0.1%三氟乙酸-乙腈,流动相梯度见下表;流速:1.0ml/min;紫外检测波长220nm;柱温:35℃;样品浓度:1mg/ml(甲醇);进样体积;5μl,化合物ⅰ保留时间r.t.=5.7min。
[0094]
流动相梯度:
[0095]
时间(min)a%b%09552030702559525.195530955
[0096]
实施例4(1r,2r)-2-(3,4-二氟苯基)环丙基甲酰胺(ⅰ)的制备
[0097][0098]
1l三口烧瓶中依次加入乙酰胺200ml,甲醇钠42.2g,化合物
ⅴ
(粗品123.6g)。机械搅拌,升温至t=60℃反应5h。原料反应完全后将搅拌下滴加水600ml,析出类白色固体。滴加完毕后缓慢降温冷却至t=0-5℃析晶3h。过滤,滤饼用纯化水(200ml)洗涤。固体于50℃鼓风干燥箱中干燥至恒重,得类白色固体化合物ⅰ(86.2g,化学纯度98.8%,收率84.0%,收率以化合物ⅱ计),
[0099]
实施例5(1r,2r)-2-(3,4-二氟苯基)环丙胺的制备
[0100][0101]
500ml三口烧瓶中加入30%氢氧化钠溶液(质量分数)243.6g。控制反应温度低于20℃下滴加12%的次氯酸钠溶液183.0g,滴加完毕后搅拌15min。将原料化合物i(60g)加入反应瓶中,t=40℃反应3h,hplc监控消失氯代中间体基本反应完全。将反应液冷却至5-10℃,浓盐酸(约170g)调反应液ph 7-9。加入乙酸乙酯300ml萃取反应液,分出上层,水层再次用乙酸乙酯150ml萃取一次,合并有机层。有机层用饱和氯化钠水洗一次(分出有机层,加入无水硫酸钠干燥。过滤得ea母液,减压浓缩至约100ml母液,待结晶用,母液hplc纯度89.5%。ms(esi)[m+1]
+
170.1.
[0102]
实施例6(1r,2s)-2-(3,4-二氟苯基)环丙胺
·
hcl的制备
[0103]
将上述实施例5制备得到的ea母液转移至三口烧瓶中,2小时滴加完hcl-ea溶液90ml(购买得到约3mol/l),滴加至结晶溶液ph=1-3(结晶温度t=15-30),继续搅拌结晶2h抽滤得类白色固体,摩尔收率80%(收率以化合物i计),m=50.1g,化学纯度99.9%。
[0104]
实施例7(1r,2s)-2-(3,4-二氟苯基)环丙胺
·
d-扁桃酸盐的制备
[0105]
将实施例5中的上述萃取液的ea母液浓缩至干得到黄色油状物,加入异丙醇100ml溶清。扁桃酸(46.3g,1eq)于异丙醇(150ml)溶清后滴加入化合物
ⅵ
的异丙醇溶液中,滴加温度t=45℃。滴加完毕后冷却至室温,析出大量白色固体,摩尔收率78%(收率以化合物i计),m=76.3g,化学纯度99.9%。
[0106]
对比例1(参照cn106906249a的实施例11)
[0107]
向350ml耐压瓶中顺序加入40g化合物v,20%氨气甲醇溶液160ml,30%甲醇钠甲醇溶液72ml,拧紧瓶盖后升温至70℃反应12h后的hplc检测图谱如图1(hplc测试条件同实施例3中的条件):
[0108]
将所得的反应液经分离,(后处理同实施例4,水解杂质成钠盐溶于结晶母液,产物酰胺不能成盐直接析出固体,达到分离的效果)将上述图谱中的两化合物分别分离得化合物(第一个峰)和(第二个峰),两化合物的鉴定数据如下所示:
[0109]
其中的核磁数据如下所示:1h nmr(dmso-d6)δ:8.81(s,3h),7.40~6.96(m,3h),2.84~2.71(m,1h),2.42(ddd,j=9.9,6.3,3.6hz,1h),1.51~1.41(m,1h),1.25~1.13(m,1h)。
13
c nmr(100hz,dmso-d6)δ:13.29,19.97,30.57,115.28,117.25,123.01,137.1,146,149,150.
[0110]
的核磁数据如下所示:1h nmr(cdcl3)δ0.92~0.98(m,1h),1.01~1.24(m,1h),1.67~1.75(m,1h),2.12~2.25(m,1h),7.01~7.13(m,2h),7.15~7.24(m,1h).
13
c nmr(101mhz,cdcl3)δ16.61,23.20,25.91,113.65,116.51,123.52,138.50,148.20,149.53,150.52,151.39,172.33.(羧基活泼氢未出峰)
[0111]
其中,hplc的测试条件如上所述,以hplc面积归一法测得,的含量大于30%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1