一类三唑类化合物、制备方法及其医药用途与流程
一类三唑类化合物、制备方法及其医药用途
(一)技术领域
1.本发明涉及药物化学领域,具体涉及一类三唑类化合物、制备方法及其医药 用途,本发明的化合物与现有技术相比,具有显著的抗组胺活性和极低的抑制胆 碱的作用,同时herg毒性明显降低。本发明还提供了其在制备预防和治疗过敏 性疾病药物中的用途。
(二)
背景技术:
2.h1受体拮抗剂可竞争性地与h1受体结合而阻断组胺与h1受体的作用,进 而抑制组胺发挥生物学效应,起到抗过敏的作用[zolaly ma.histamine h
1 antagonists and clinical characteristics of febrile seizures[j].int j gen med.2012,5: 277-281]。但是,目前临床正在使用的第三代h1受体拮抗剂地氯雷他定等,虽 然未见明显的心脏毒性,但是存在明显的抑制胆碱的副作用,如口干,眼睛干涩, 严重会引起运动功能障碍。因此,开发新的具有更高安全性的h1受体阻断剂, 改善现有治疗药物的不足,降低中枢、心脏毒性和胆碱抑制活性,具有重要的价 值。
[0003]
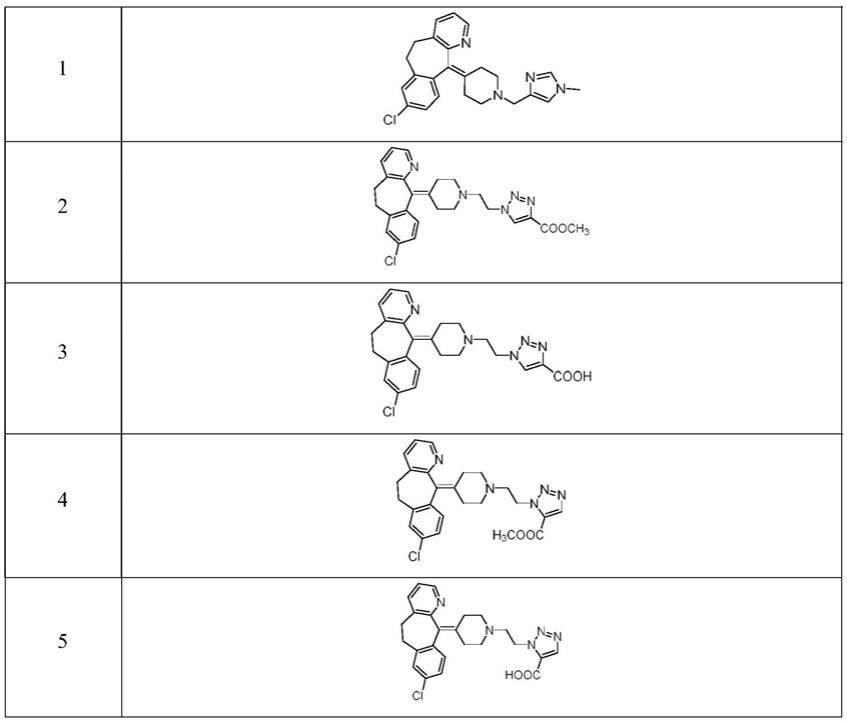
本发明人申请的中国专利cn107602534b中公开了一系列具有抗组胺和抗炎 双重活性的地氯雷他定衍生物,其中化合物lhc-7活性最佳,
[0004][0005]
但是在随后的研究中发现其具有很强的herg毒性。
[0006]
本发明人申请的中国专利cn109096251a中公开了一类多靶点地氯雷他定 衍生物,包括化合物lp-2。
[0007][0008]
专利wo9200293a1中也公开了一系列的抗组胺化合物,其中包括以下结构 的化合物1:
[0009][0010]
但该专利中未公开化合物1的实施方法、活性数据等。
[0011]
经过不断努力,本发明设计了一系列新的化合物,并发现这些化合物表现出 优异的效果和作用。尤其是本发明化合物既保留了显著的抗组胺的活性,同时明 显降低其抑制m胆碱的能力,具有更高的安全性。
(三)
技术实现要素:
[0012]
本发明针对现有技术的不足,通过结构改造,提供一种具有下列式(i)的化合 物或其药用盐
[0013][0014]
其中
[0015]
a环为苯环、吡啶环、噻吩环;
[0016]
x为被r5、r6取代或未取代的二唑,三唑,四唑,噁唑酮,噁二唑酮,1,2,4-三 唑-3-酮;
[0017]
n=1-6;
[0018]
r1、r4选自h、c1~c6的烷基、羟基、卤素;
[0019]
r2、r3选自h、c1~c6的烷基、羟基、=o;
[0020]
r5、r6选自h、c1~c6的烷基、coor7、conr7、 r7为h,任意取代的c1~c6的烷基。
[0021]
优选的,式(i)化合物
[0022]
其中
[0023]
n=1-4;
[0024]
r1、选自h、cl;
[0025]
r2、r3、选自h、羟基;
[0026]
r4为h;
[0027]
r5、r6选自h、甲基、coor7、conr7、
[0028]
r7为h,甲基。
[0029]
更优选的,式(i)化合物
[0030]
其中
[0031]
a环为吡啶环。
[0032]
本发明进一步包括本发明化合物的药用盐,所述药用盐为式(i)化合物和碱金 属或者碱土金属,氨基酸或者含有氨基的碱性化合物形成的盐,或者为式(i) 化合物与无机酸或者有机酸形成的盐,或者为式(i)化合物多元酸碱金属或者 碱土金属盐复合盐。
[0033]
具体的,包括式(i)化合物的钾盐,钠盐,铵盐以及式(i)化合物与盐酸、硫 酸、磷酸、氢溴酸、马来酸、富马酸、枸橼酸、甲磺酸、对甲苯磺酸、酒石酸或 醋酸形成的盐;所述式(i)化合物多元酸碱金属或者碱土金属盐复合盐中,其 中的多元酸,选自枸橼酸、丁二酸、酒石酸、琥珀酸、富马酸、马来酸、草酸、 硫酸、磷酸、亚硫酸、苹果酸,其中的碱金属或碱土金属选自钠、钾、钙、镁、 锌。
[0034]
特别优选的本发明化合物,选自以下化合物:
[0035]
[0036]
[0037][0038]
本发明进一步包括含有本发明化合物或其药用盐的药物组合物。其剂型选自片剂, 胶囊剂、颗粒剂、口服液、栓剂、透皮制剂、注射剂。
[0039]
本发明进一步包括本发明的化合物或其药用盐在制备预防和治疗过敏性疾病的 药物中的用途。其中所述过敏性疾病选自:过敏性鼻炎、荨麻疹、慢性荨麻疹、 过敏性紫癜、哮喘、过敏性皮炎、湿疹、过敏性结膜炎、特应性皮炎、鼻窦炎、 慢性鼻窦炎。
[0040]
药效学实验表明本发明的化合物显著的拮抗组胺h1受体活性,极显著的降低或 者基本消除了现有技术中公开的化合物的抗胆碱活性,降低了化合物herg毒性, 本发明还包括本发明化合物的前药、互变异构体、立体异构体或者立体异构体的 混合物。
[0041]
(四)生物活性
[0042]
1.本发明化合物对组胺诱导的小鼠血管通透性的抑制作用
[0043]
取icr小鼠,体重18-22g,适应性饲养7天,随机分组,每组10只,即模 型组、本发明化合物组、阳性药地氯雷他定组、lhc-7对照组、化合物1对照组。 于造模前1h灌胃给药,对照组给予相应溶媒,化合物组给予5mg/kg相应化合物, 地氯雷他定组分别给予5mg/kg相应药物。小鼠用10%硫化钠将小鼠背部脱毛 (3cm
×
3cm),给药1h后于脱毛处皮内注射磷酸组胺生理盐水溶液1ug/0.1ml,注 射后立即由尾静脉注射0.25%伊文思蓝溶液4ml/kg,30min后脱臼处死,取下 蓝染部分的皮肤打孔(直径15mm),剪碎后浸泡于丙酮-生理盐水(7:3)溶液1ml中, 24h后离心(3000r/min,10min),取上清液,用紫外分光光度计在波长610nm处, 以丙酮-生理盐水溶液空白校零,测定所取样本的吸光度值。
[0044]
结果见表1,与模型组比较,各化合物组及地氯雷他定组均具有显著的抑制 组胺诱导小鼠皮肤血管通透性(p<0.01);与对照组比较,本发明化合物抑制率与 地氯雷他定、lhc-7、lp-2化合物抑制率相当,优于化合物1(p<0.05)。说明, 本发明化合物具有较强的抗组胺活性。
[0045]
表1本发明化合物对组胺诱导的小鼠血管通透性的抑制作用
[0046][0047][0048]
2.本发明化合物对毛果芸香碱诱导的大鼠流涎的试验结果
[0049]
wistar雄性大鼠,180-220g,分为空白组、模型组、地氯雷他定组、本发明 化合物组,lhc-7对照组、化合物1对照组、每组10只。各组大鼠灌胃给予相 应药物,15min后腹腔注射毛果芸香碱(1mg/kg,生理盐水溶解)诱导唾液分泌。 立即腹腔注射10%水合氯醛麻醉大鼠,大鼠麻醉后,口腔内残留的唾液用棉球取 出,然后将称重过(m1)的棉球放入口腔内(舌下两个,两侧各一个),10分 钟后取出立即测量棉球重量(m2)。
[0050]
唾液分泌量=m2-m1
[0051]
抑制率=给药组唾液分泌量/模型组唾液分泌量
×
100%
[0052]
实验结果如下表所示,模型组腹腔注射毛果芸香碱1mg/kg,唾液分泌量显 著增
加,模型成功。灌胃给药地氯雷他定(1mg/kg),lhc-1(10mg/kg)、lp-2 (10mg/kg)、化合物1(10mg/kg)唾液分泌量减少(p<0.01),说明地氯雷他 定、lhc-1、lp-2和化合物1有明显的抑制胆碱的作用。灌胃给药本发明化合物 (10mg/kg)唾液分泌量与模型组比较无统计学差异(p>0.05),与对照组比较 具有显著性差异(p<0.05)。说明与地氯雷他定、lhc-7、lp-2和化合物1相比, 本发明化合物抑制胆碱的活性极低。
[0053]
表2本发明化合物对毛果芸香碱诱导的大鼠流涎的抑制作用
[0054][0055][0056]
3.本发明化合物herg毒性测试
[0057]
本发明化合物与对比技术化合物lhc-7、lp-2、化合物1及地氯雷他定相 比,herg毒性明显降低,特别是实施例3、5、7、13、19、22、23、25抑制herg 的ic
50
>30μm,具有更高的安全性。
[0058]
实施例编号herg(μm)16.511215.2433>305>307>30815.32313>3019>3022>3023>3025>30
lhc-70.248lp-24.032化合物12.033地氯雷他定1.214
[0059]
(五)具体实施例方式
[0060]
实施例1
[0061]
8-氯-11-(1
–
((5-甲基-1h-咪唑-4-基)甲基)哌啶-4-亚基)-6,11-二氢-5h-苯并[5,6] 环庚[1,22-b]吡啶的合成
[0062]
化合物地氯雷他定(310mg,1mmol)、5-甲基-1h-咪唑-4-甲醛(330mg,3 mmol)溶于1.2-二氯乙烷(50ml)中,室温搅拌反应10min,然后加三乙酰氧基硼 氢化钠(626mg,3mmol),醋酸(3滴)。升温至25℃搅拌反应过夜。lc(dcm:meoh =10:1)检测原料反应完全。浓缩,拌样,柱层析得目标化合物(190mg,47%yield), 白色固体。1h nmr(400mhz,cdcl3):δppm 8.36(dd,j1=1.2hz,j2=4.8hz,1h), 7.74(s,1h),7.47(dd,j1=0.8hz,j2=7.8hz,1h),7.47(d,j=2.0hz,1h),7.10-7.14 (m,2h),7.04(d,j=8.0hz,1h),4.09(s,2h),3.31-3.39(m,2h),3.06-3.20(m,4h), 2.61-2.89(m,6h),2.28(s,3h).
[0063]
实施例2
[0064]
步骤1 2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)乙 烷-1-醇(2)的合成
[0065][0066]
将原料1(400mg,1.29mmol)和n,n-二异丙基乙胺(417mg,3.23mmol) 溶于dcm(7ml),室温搅拌30min后,加入2-溴乙醇(404mg,3.23mmol), 室温搅拌反应22h。tlc(v丙酮:v二氯甲烷:v三乙胺=1:2:0.1)检测原料1反应 完全。停止反应,减压旋干溶剂,柱层析(v二氯甲烷:v乙酸乙酯:v三乙胺=2:1: 0.05)分离,得白色固体412mg,产率90.0%。1h-nmr(300mhz,cdcl3)δ(ppm): 8.42(d,j=4.2hz,1h,arh),7.46(d,j=7.5hz,1h,arh),7.19-7.12(m,4h,arh), 3.78-3.75(m,2h,ch2oh),3.45-3.34(m,2h,arch2),2.97-2.57(m,12h,arch2, n(ch2)3,c(ch2)2).
[0067]
步骤2
[0068]
2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)甲磺酸乙 酯(3)的合成
[0069][0070]
将2(520mg,1.47mmol)溶于二氯甲烷(8ml)中,于0℃下滴加入甲磺 酰氯(253mg,2.21mmol),0℃搅拌反应1h。tlc(v二氯甲烷:v甲醇=20:1) 检测原料2反应完全后,向反应液中加入饱和氯化铵溶液(10ml)搅拌5min 后分液,水层继续用二氯甲烷(10mlx3)萃取,合并有机层用无水硫酸钠干燥, 抽滤,减压旋干溶剂,不经纯化直接投下一步。
[0071]
步骤3
[0072]
11-(1-(2-叠氮乙基)哌啶-4-亚基)-8-6,11-二氢-5h-苯并[5,6]环庚[1,2-b]吡啶(4) 的合成
[0073][0074]
将3(636mg,1.47mmol)溶于dmf(5ml)中,冰浴冷却至0℃,分批加 入叠氮化钠(191mg,2.94mmol),加毕低温搅拌10min,加热至50℃反应12 h。tlc(v二氯甲烷:v甲醇=20:1)检测原料3反应完全。停止加热,冷却至室 温,加入饱和次氯酸钠溶液(5ml)淬灭,加入乙酸乙酯萃取,水层再用乙酸 乙酯萃取(10mlx3),合并有机层,加无水硫酸钠干燥,抽滤,减压旋干滤液, 柱层析(洗脱剂:v二氯甲烷:v甲醇=200:1~50:1)得白色固体370mg,产率 66.3%。
[0075]1h-nmr(300mhz,cdcl3)δ(ppm):8.42(d,j=4.5hz,1h,arh),7.46(d,j=7.6 hz,1h,arh),7.18-7.10(m,4h,arh),3.46-3.38(m,2h,arch2),2.89-2.35(m,12h, arch2,n(ch2)3,c(ch2)2),1.88-1.78(m,2h,ch2n3).
[0076]
步骤4
[0077]
1-(2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)乙基)
ꢀ-
1h-1,2,3-三唑-4-甲酸甲酯的合成
[0078][0079]
将4(500mg,1.32mmol)溶于乙腈和水的混合溶液中(v乙腈:v水=6:1, 8ml),于冰浴下依次加入9(130mg,1.55mmol)和碘化亚铜(50mg,0.26mmol), 加毕低温搅拌30min后移入室温下反应2h。tlc(v二氯甲烷:v甲醇=20:1) 检测原料4反应完全,停止反应,减压旋干溶剂,加甲醇(5ml)打浆,抽滤, 固体再用二氯甲烷(5ml)打浆,抽滤,合并滤液,减压旋干溶剂,残留物经 柱层析(洗脱剂:v二氯甲烷:v甲醇=200:1~50:1)分离,得淡灰黄色固体 298mg,产率:48.7%,m.p.185.8~187.5℃。1h-nmr(300mhz,cdcl3)δ(ppm): 8.45-8.44(m,1h,arh),8.28(s,1h,arh),7.51(d,j=7.2hz,1h,arh),7.16-7.13(m, 4h,arh),4.52(t,j=6.0hz,1h,triazole-ch2),3.98(s,3h,och3),3.41-3.35(m,2h, arh-ch2ch2),2.87-2.79(m,6h,arh-ch2ch2,n(ch2)2),2.56-2.31(m,6h,nch2, c(ch2)2).
[0080]
实施例3
[0081]
1-(2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)乙 基)-1h-1,2,3-三唑-5-甲酸甲酯的合成
[0082]
参考实施例2的合成方法,柱层析(洗脱剂:v二氯甲烷:v甲醇=200:1~50: 1)分离,得浅棕色固体210mg化合物,收率:43.1%,m.p.186.2~188.1℃。
[0083]
实施例4
[0084]
1-(2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)乙基)
ꢀ-
1h-1,2,3-三唑-4-甲酸的合成
[0085]
将实施例2化合物(100mg,0.22mmol)溶于四氢呋喃(2ml),冰浴下滴 加lioh.h2o的水溶液(23mg,2ml),加毕低温搅拌10min后移入室温下反 应1h。tlc(v二氯甲烷:v甲醇=30:1)检测原料dl-1反应完全,减压旋干 溶剂,加1ml水搅拌溶解,冰浴下用6n hcl调ph=4~5,析出固体,抽滤烘干 得白色固体62mg,收率:62.6%,m.p.80.1~82.0℃。
[0086]
实施例5
[0087]
1-(2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)乙 基)-1h-1,2,3-三唑-5-甲酸的合成
[0088]
以实施例3(200mg,0.44mmol)原料,操作同实施例4的合成方法,得白 色固体121mg,收率:61.1%,m.p.>250℃。
[0089]
实施例6
[0090]
参考实施例4的合成方法,制得化合物64mg,收率34.1%,1h-nmr(300mhz, cd3od)δ(ppm):2.44~2.55(m,2h),2.64~2.66(m,2h),2.82~2.92(m,2h),3.04~ 3.06(m,2h),3.35~3.37(m,2h),3.39~3.42(m,2h),4.45(s,2h),5.48(s,1h),7.11(d, 1h),7.19(d,1h),7.25(s,1h),7.27(t,1h),7.65(d,1h),8.33(d,1h).
[0091]
实施例7
[0092]
参考实施例4的合成方法,制得化合物58mg,收率26.3%,1h-nmr(300mhz, cd3od)δ(ppm):2.17~2.23(m,2h),2.26~2.35(m,2h),2.39~2.44(m,2h), 2.78~2.81(m,2h),2.84~2.90(m,4h),3.37~3.42(m,2h),3.58~3.60(m,2h), 3.65~3.67(m,2h),4.51~4.54(m,2h),4.64(s,2h),7.07(d,1h),7.13(d,1h),7.20(s, 1h),7.25(t,1h),7.65(d,1h),8.03(s,1h),8.32(s,1h).
[0093]
实施例8
[0094]
1-(2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)乙 基)-1h-1,2,3-三唑-4-甲酰胺的合成
[0095]
将实施例2化合物(50mg,0.11mmol)溶于氨水(2ml)中,封管78℃反 应3h。冷却至室温,tlc(v二氯甲烷:v甲醇=20:1)检测原料dl-1反应完 全,反应液抽滤,白色固体经水洗,烘干,得30mg,收率:60.7%,m.p.234.5~236.1℃。
[0096]1h-nmr(300mhz,cdcl3)δ(ppm):8.43-8.42(m,1h,arh),8.26(s,1h,arh), 7.46(d,j=6.8hz,1h,arh),7.18-7.09(m,4h,arh),4.53(t,j=6.6hz,1h, triazole-ch2),3.46-3.34(m,2h,arh-ch2ch2),2.87-2.74(m,6h,arh-ch2ch2, n(ch2)2),2.56-2.32(m,6h,nch2,c(ch2)2).
[0097]
实施例9
[0098]
1-(2-(4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)乙 基)-1h-1,2,3-三唑-5-甲酰胺的合成
[0099]
以实施例3化合物(100mg,0.22mmol)原料,操作同实施例8,得灰白色 固体70mg,收率:70.9%,m.p.220.1~221.8℃。
[0100]
1h-nmr(300mhz,cdcl3)δ(ppm):8.43-8.41(m,1h,arh),8.26(s,1h,arh), 7.46(d,j=8.1hz,1h,arh),7.18-7.10(m,4h,arh),4.53(t,j=6.3hz,1h, triazole-ch2),3.46-3.35(m,2h,arh-ch2ch2),2.88-2.73(m,6h,arh-ch2ch2, n(ch2)2),2.60-2.30(m,6h,nch2,c(ch2)2).
[0101]
实施例10
[0102]
步骤1
[0103][0104]
向500ml单口瓶中依次加入地氯雷他定(8.00g,25.7mmol)、碳酸钾(7.12g, 51.5mmol)、丙酮(200ml)和1-溴-2-氯乙烷(9.67g,51.5mmol),氮气保护下,室 温反应过夜。tlc(dcm:meoh=10:1)监测反应结束后,浓缩。乙酸乙酯(100ml) 加入残留物中,过滤、浓缩并经柱层析纯化(dcm:meoh=100:1~25:1)得到中间 体1(2.60g,27.1%yield),淡红色固体。1h nmr(400mhz,cdcl3):δppm 8.39-8.41(dd,j1=1.6hz,j2=4.8hz,1h),7.42-7.44(m,1h),7.14-7.15(m,1h), 7.13(d,j=1.2hz,2h),7.07-7.10(m,1h),3.58(t,j=7.2hz,2h),3.32-3.44(m,2 h),2.76-2.87(m,4h),2.73(t,j=7.2hz,2h),2.50-2.57(m,
1h),2.40-2.47(m,1 h),2.23-2.39(m,4h).
[0105]
步骤2
[0106][0107]
向100ml三口瓶中依次加入2,2-二甲基丁-3-炔酸(2.00g,17.8mmol)、二氯甲 烷(40ml)、dmap(261mg,2.14mmol)、dcc(4.41g,21.4mmol)和苯甲醇(2.31g, 21.4mmol),氮气保护下,室温反应过夜。tlc(pe:ea=20:1)监测反应结束后, 反应液过滤,浓缩,并经快速制备色谱纯化(40g,pe:ea=200:1)得到中间体2(3.25 g,90.0%yield),浅黄色油状物。1h nmr(400mhz,cdcl3):δppm 7.33-7.38(m,5 h),5.20(s,2h),2.29(s,1h),1.53(s,6h).
[0108]
步骤3
[0109][0110]
向100ml三口瓶中依次加入中间体2(2.00g,9.89mmol)、(dmf:meoh=9:1,40 ml)、cui(94mg,0.49mmol)和tmsn3(1.71g,14.8mmol),氮气保护下,缓慢升 温至100℃,保温反应过夜。反应液冷却至室温,tlc(pe:ea=20:1)监测反应结 束。水(120ml)淬灭反应体系,乙酸乙酯(30ml
×
6)萃取,饱和氯化钠溶液(30 ml
×
3)洗涤,收集有机相,无水硫酸钠干燥。过滤、浓缩并经快速制备色谱纯化 (40g,pe:ea=4:1)得到中间体3(1.87g,76.9%yield),无色油状物。1h nmr(400 mhz,dmso-d6):δppm 14.80(s,1h),7.76(s,1h),7.24-7.36(m,5h),5.10(s,2 h),1.56(s,6h).
[0111]
步骤4
[0112][0113]
向100ml三口瓶中加入中间体3(1.60g,6.52mmol)和无水四氢呋喃(24ml),氮 气保护下,降温至0℃。分三批向上述反应液中加入氢化钠(60%,391mg,9.78 mmol),保温搅拌30min后,再缓慢滴加2-(三甲基硅烷基)乙氧甲基氯(1.63g,9.78 mmol),加毕,室温反应8h。tlc(pe:ea=4:1)监测反应结束后,反应液降温至0℃。 水(100ml)淬灭,乙酸乙酯(20ml
×
3)萃取,饱和氯化钠溶液(20ml
×
3)洗涤,收 集有机相,无水硫酸钠干燥。过滤,浓缩得到中间体4(2.45g,100%yield,n-位 置异构的混合物),油状物,直接投入下步使用。1h nmr(400mhz,cdcl3):δppm 7.56(s,1h),7.25-7.36(s,5h),5.61(s,2h),5.13(s,2h),3.60-3.64(m,2h),1.64 (m,6h),0.87-0.92(m,2h),-0.04(s,9h).(ywf41-12-1,20190606).1h nmr(400 mhz,cdcl3):δppm 7.56(s,1h),7.27-7.36(s,5h),5.61(s,2h),5.14(s,2h),
[0114]
3.55-3.59(s,2h),1.68(s,6h),0.87-0.91(s,2h),-0.03(s,9h).
[0115]
步骤5
[0116][0117]
向250ml单口瓶中依次加入4(2.45g,6.52mmol)、甲醇(50ml)和 10%pd/c(0.98g,40%wt),氢气氛围下,45℃反应过夜(15psi)。tlc(pe:ea=7.5:1) 监测反应结束后,硅藻土铺垫下抽滤,浓缩得到5(1.86g,100%yield,位置异构 的混合物),淡黄色油状物,直接投入下一步。
[0118]
步骤6
[0119][0120]
向100ml单口瓶中加入5(1.86g,6.52mmol)和(dcm:meoh=10:1,44ml), 0℃条件下缓慢滴加三甲基硅重氮甲烷(3.6ml,7.17mmol),加毕,室温反应4h。 tlc(pe:ea=7.5:1)监测、溴甲酚绿显色显示反应结束后,浓缩并经快速制备色谱 纯化(40g,pe:ea=20:1~10:1)得到6(630mg,70%purity),无色油状物。1h nmr (400mhz,cdcl3):δppm 7.60(s,1h),5.63(s,2h),3.69(s,3h),3.57-3.61(s,2h), 1.66(s,6h),0.88-0.92(s,2h),-0.03(s,9h).
[0121]
步骤7
[0122][0123]
向100ml单口瓶中依次加入6(893mg,2.98mmol)、四氢呋喃(31ml)和四 丁基氟化铵(3.89g,14.9mmol),氮气保护下,66℃反应4h。tl c(pe:ea=2:1) 监测反应结束后,浓缩。残留物中加入乙酸乙酯(40ml),饱和氯化钠溶液(40 ml
×
6)洗涤,收集有机相,无水硫酸钠干燥。过滤、浓缩并经快速制备色谱纯化 (12g,pe:ea=2:1)得到7(400mg,79.3%yield),无色油状物。1h nmr(400mhz, cdcl3):δppm 7.65(s,1h),3.71(s,3h),1.66(s,6h).
[0124]
步骤8
[0125][0126]
向100ml单口瓶中依次加入7(250mg,1.48mmol)、dmf(16ml)、碳酸钾 (409mg,2.96mmol)、1(552mg,1.48mmol)和碘化钾(25mg,0.15mmol),氮气保 护下,70℃反应4h。tlc(dcm:meoh=20:1)监测反应结束后,0℃条件下, 水(90ml)淬灭反应体系。乙酸乙酯(10ml
×
6)萃取、饱和氯化钠溶液(20ml
×
3) 洗涤,收集有机相,无水硫酸钠干燥。过滤、浓缩并经柱层析(ea:meoh=25:1) 得到实施例10化合物(497mg,41.4%yield),黄色油状物。1h nmr
(400mhz, cdcl3):δppm 8.38-8.40(dd,j1=1.6hz,j2=4.8hz,1h),7.46(s,1h),7.41-7.43(dd, j1=1.6hz,j2=7.6hz,1h),7.14(s,1h),7.10-7.12(m,2h),7.06-7.09(m,1h),4.50 (t,j=7.2hz,2h),3.65(s,3h),3.31-3.46(m,2h),2.93(t,j=6.8hz,2h),2.71-2.86 (m,4h),2.45-2.52(m,1h),2.27-2.41(m,3h),2.18-2.25(m,2h),1.59(s,6h).
[0127]
实施例11
[0128]
参考实施例10的合成方法,制得实施例11化合物(172mg,14.3%yield),黄 色油状物。1h nmr(400mhz,cdcl3):δppm 8.39-8.40(dd,j1=1.6hz,j2=4.8hz, 1h),7.50(s,1h),7.42-7.44(dd,j1=1.6hz,j2=7.6hz,1h),7.15(br,1h),7.12(br, 2h),7.07-7.10(m,1h),4.21(t,j=7.6hz,2h),3.71(s,3h),3.32-3.44(m,2h), 2.94(t,j=7.6hz,2h),2.76-2.87(m,4h),2.47-2.54(m,1h),2.31-2.44(m,3h), 2.20-2.29(m,2h),1.63(s,6h).
[0129]
实施例12
[0130]
参考实施例10的合成方法,制得实施例12化合物(48mg,4.00%yield),黄 色油状物。1h nmr(400mhz,cdcl3):δppm 8.38-8.40(dd,j1=1.6hz,j2=4.8hz, 1h),7.59(s,1h),7.42-7.44(dd,j1=1.6hz,j2=7.6hz,1h),7.15(br,1h),7.12(br, 2h),7.07-7.10(m,1h),4.43(t,j=6.4hz,2h),3.67(s,3h),3.31-3.44(m,2h), 2.73-2.87(m,6h),2.48-2.55(m,1h),2.31-2.44(m,3h),2.21-2.31(m,2h),1.63(s, 6h).
[0131]
实施例13
[0132][0133]
向25ml单口瓶中依次加入实施例10化合物(120mg,0.24mmol)、甲醇(4 ml)和氢氧化锂单水合物(40mg,0.95mmol)的水溶液(1ml),氮气保护下,室温 反应过夜。tlc(ea:meoh=20:1)监测反应结束后,浓缩,除去有机溶剂。水(1ml) 稀释残留液,1n盐酸调节ph(5),析出大量固体,室温搅拌1h。过滤、滤饼以 水(2ml
×
4)洗涤,收集滤饼,鼓风干燥(50℃,4h)得到实施例13化合物(100mg, 84.7%yield),白色固体。1h nmr(400mhz,cdcl3):δppm 8.47-8.49(dd,j1=1.2 hz,j2=4.8hz,1h),7.56(s,1h),7.52-7.54(dd,j1=0.8hz,j2=7.6hz,1h), 7.16-7.19(m,3h),7.10(s,1h),4.82-4.90(m,1h),4.36-4.41(m,1h),3.24-3.36 (m,4h),3.13(t,j=10.0hz,1h),2.75-2.81(m,3h),2.53-2.57(m,1h),2.22-2.29 (m,1h),2.01-2.10(m,2h),1.78-1.89(m,2h),1.72(s,3h),1.64(s,3h). (ywf42-01-1,20190622).
13
c nmr(100mhz,cdcl3):δ177.81,156.29,152.26, 144.66,138.46,137.82,137.55,136.50,133.68,132.48,131.58,130.80,130.57, 128.73,125.74,122.22,55.49,51.32,50.76,50.49,41.22,31.17,30.35,29.77,24.93, 24.28.
[0134]
实施例14
[0135]
参考实施例13的合成方法,制得实施例14化合物(110mg,65.8%yield),白 色固体。1h nmr(400mhz,cdcl3):δppm 8.47-8.49(dd,j1=1.6hz,j2=4.8hz,1 h),7.74(s,1h),7.48-7.50(dd,j1=1.6hz,j2=7.6hz,1h),7.11-7.16(m,3h), 7.05-7.07(d,j=8.4hz,
1h),4.45-4.57(m,2h),3.31-3.41(m,2h),2.97-3.09(m,2 h),2.75-2.86(m,3h),2.53-2.57(m,1h),2.27-2.50(m,5h),2.16-2.21(m,1h), 1.69(s,3h),1.66(s,3h).
13
c nmr(100mhz,cdcl3):δ178.66,156.57,152.44, 145.84,139.32,138.13,137.35,137.24,133.83,133.00,132.47,130.63,129.06, 126.18,122.60,122.13,56.56,53.81,53.75,47.15,42.01,31.61,31.22,29.99,29.90, 26.27,25.82。
[0136]
实施例15
[0137]
参考实施例13的合成方法,制得实施例15化合物(83mg,94.3%yield),类 白色固体。1h nmr(400mhz,cdcl3):δppm 8.35(d,j=4.0hz,1h),7.52(s,1h), 7.46(d,j=7.2hz,1h),7.09-7.16(m,3h),6.98(d,j=8.4hz,1h),4.55-4.69(m,2 h),3.69-3.76(m,1h),3.55-3.61(m,1h),3.29-3.39(m,2h),3.15-3.18(m,1h), 2.74-2.89(m,5h),2.47-2.59(m,2h),2.28-2.40(m,2h),1.64(s,3h),1.61(s,3h). 13
c nmr(100mhz,cdc3):δ174.56,151.54,141.90,139.03,135.10,133.67, 132.57,130.15,129.95,129.18,128.88,127.28,125.67,124.61,121.90,118.23, 50.31,49.22,49.06,40.36,37.32,27.05,27.00,24.15,23.88,22.20,22.14。
[0138]
实施例16-22
[0139]
参考实施例13的合成方法,制得实施例16-22化合物,具体如下
[0140]
[0141][0142]
实施例23
[0143]
5-((4-(8-氯-5,6-二氢-11h-苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-基)甲 基)-2.4-二氢-3h-1,2,4-三唑-3-酮的合成
[0144][0145]
将地氯雷他定(500mg,1.6mmol)和diea(520mg,4.0mmol)溶于甲醇 (8ml),于室温下搅拌30min后缓慢滴加入5-(氯甲基)-2,4-二氢-3h-1,2,4
-ꢀ
三唑-3-酮(215mg,1.6mmol),滴加完毕后室温反应4h。tlc(v二氯甲烷: v甲醇=15:1)检测原料1反应完全,停止反应,补加甲醇(10ml)搅拌30min 后,抽滤,烘干得白色固体402mg,收率:63.4%,m.p.>250℃。
[0146]1h-nmr(300mhz,cdcl3)δ(ppm):8.43-8.41(m,1h,arh),7.47(d,j=8.4hz, 1h,arh),7.19-7.14(m,4h,arh),3.59(s,1h,nch2),3.43-3.36(m,2h, arh-ch2ch2),2.95-2.81(m,4h,arh-ch2ch2,n(ch2)2),2.70-2.45(m,6h,n(ch2)2, c(ch2)2).
[0147]
实施例24
[0148]
参考实施例23的合成方法,制得化合物123mg,收率46.1%,1h-nmr(300 mhz,cd3od)δ(ppm):2.24~2.34(m,2h),2.44~2.50(m,2h),2.83~2.85(m, 2h),2.86~2.92(m,2h),3.21~3.25(m,2h),3.34(s,3h),3.54(s,2h),3.70~3.76(m,2h), 7.11(d,1h),7.16(d,1h),7.22(s,1h),7.27(q,1h),7.68(d,1h),8.32(d,1h).
[0149]
实施例25
[0150]
参考实施例23的合成方法,制得化合物136mg,收率38.3%,1h-nmr(300 mhz,cd3od)δ(ppm):2.31~2.39(m,2h),2.41~2.45(m,2h),2.49~2.52(m,2h), 2.83~2.84(m,2h),2.90~2.92(m,2h),3.45~3.47(m,2h),3.69(s,2h),7.16(d,1h), 7.21(d,1h),7.27(s,1h),7.29(q,1h),7.70(d,1h),8.36(d,1h)。
[0151]
本发明专利的组合物实施例,以实施例33化合物为例,包含但不限于下列实施 例中使用的辅料及处方配比。
[0152]
实施例26
[0153]
实施例1化合物包衣片(1000片)
[0154]
片芯处方
[0155][0156]
包衣液处方
[0157][0158]
制备方法:实施例1化合物、硬脂酸镁过120目筛,微晶纤维素、磷酸氢钙、 预胶化淀粉过100目筛,备用;称取处方配比量的实施例1化合物、微晶纤维素、 磷酸氢钙、预胶化淀粉,按等量递增法过80目筛混匀,用30%的乙醇溶液作为 润湿剂制软材,20目筛制粒,50~60℃干燥3~4小时,20目筛整粒,加入处方量 的硬脂酸镁,混匀,压片制得。
[0159]
实施例27
[0160]
实施例1化合物的普通片(1000片)
[0161][0162]
制备方法:实施例1化合物、硬脂酸镁过120目筛,微晶纤维素、羧甲基淀 粉钠、乳糖过80目筛,备用;称取处方配比量的实施例1化合物、微晶纤维素、 羧甲基淀粉钠和乳糖,按等量递增法过80目筛混匀,用30%的乙醇溶液作为润 湿剂制软材,30目筛制粒,50~60℃干燥3~4小时,30目筛整粒,加入处方量的 硬脂酸镁,混匀,压片制得。
[0163]
实施例28
[0164]
实施例1化合物的胶囊(1000粒)
[0165][0166]
制备方法:实施例1化合物、硬脂酸镁过120目筛,低取代羟丙基纤维素、 羧甲基淀粉钠、乳糖过80目筛,备用;称取处方配比量的实施例1化合物、低 取代羟丙基纤维素、羧甲基淀粉钠和乳糖,按等量递增法过80目筛混匀,用30% 的乙醇溶液作为润湿剂制软材,30目筛制粒,50~60℃干燥3~4小时,30目筛整 粒,加入处方量的硬脂酸镁,混匀,灌装胶囊即可。
[0167]
实施例29
[0168]
实施例1化合物的颗粒剂(1000包)
[0169][0170]
制备方法:实施例1化合物、微晶纤维素、蔗糖粉、羧甲基淀粉钠、乳糖、 阿斯巴甜过100目筛,橘子香精、十二烷基硫酸钠过80目筛,备用;称取处方 配比量的实施例1化合物、微晶纤维素、蔗糖粉、羧甲基淀粉钠、乳糖、阿斯巴 甜按等量递增法混匀,用3%聚维酮的30%乙醇溶液制软材,20目筛制粒,50~60℃ 干燥3~4小时,18目筛整粒,加入处方量的橘子香精、十二烷基硫酸钠,混匀, 装袋封口制得。
[0171]
实施例30
[0172]
实施例1化合物口服液(1000瓶)
[0173][0174]
制备方法:阿斯巴甜、橘子香精、枸橼酸钠溶于现制注射用水中,过滤,常 温下加入处方量的实施例1化合物,溶解,过滤,灌装。
[0175]
实施例31
[0176]
实施例1化合物糖浆剂
[0177]
[0178][0179]
制备方法:将蔗糖加入900ml注射用水中,加热煮沸,溶解、趁热过滤,冷 却至室温备用;将处方量的实施例1化合物、阿斯巴甜、橘子香精、枸橼酸钠溶 于60ml注射用水中,过滤,加入至上述糖浆液中,加注射用水至1000ml,混 匀,灌装制得。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1