艾滋病病毒膜融合抑制剂脂肽及药物用途的制作方法
本申请是申请号为201710251788.7、申请日为2017年04月18日、发明创造名称为“强效抑制hiv的脂肽、其衍生物、其药物组合物及其用途”的分案申请。
本发明涉及生物医药领域中艾滋病病毒膜融合抑制剂脂肽及药物用途。
背景技术:
艾滋病(aids)是当前严重危害人类健康和社会发展的重要传染病。引起艾滋病的人免疫缺陷病毒分为hiv-1型和hiv-2两型。全球现有hiv感染人数大约3600万,hiv-1是主要的病原体(www.unaids.org)。目前尚无有效的艾滋病疫苗问世,阻断病毒不同复制阶段的药物对治疗和预防hiv感染起着重大作用。目前临床上的治疗药物主要包括核苷类逆转录酶抑制剂、非核苷类逆转录酶抑制剂、蛋白酶抑制剂、病毒进入抑制剂、整合酶抑制剂(www.fda.gov)。临床广泛使用的高效抗病毒治疗方案即所谓的“鸡尾酒”疗法主要由3-4种逆转录酶抑制剂和蛋白酶抑制剂组成。由于hiv感染的持续性,病人需要长期给药,所以容易导致耐药性的产生,严重地影响临床治疗效果[1]。因此,研发新的抗hiv药物一直是防控艾滋病的重要策略。
不同于其它类型的药物,hiv进入抑制剂作用于病毒复制的早期阶段,通过阻断病毒进入靶细胞而发挥作用,如同“拒敌人于国门之外”,因此在治疗和预防上均具有明显的优势。然而,目前仅有两种hiv进入抑制剂批准用于临床:第一个是hiv膜融合抑制剂恩夫韦肽(enfuvirtide,又名t-20),其为一来源于hiv融合蛋白gp41的具有36个氨基酸的多肽药物;第二个是辅助受体ccr5拮抗剂马拉维若(maraviroc)。它们的研发成功为艾滋病临床治疗增添了新的手段,但令人遗憾的是,t-20由于活性相对较低需要每天大剂量给药(每天两次90mg皮下注射),且容易导致耐药产生;马拉维若选择性地针对ccr5嗜性病毒,而对crcr4嗜性病毒无效[2]。
hiv进入靶细胞由其表面的包膜糖蛋白(env)介导,该蛋白由表面亚基gp120和跨膜亚基gp41通过非共价键连接而成,在天然状态下呈三聚体结构[3]。首先,gp120先后与细胞受体cd4和辅助受体(如ccr5或cxcr4)结合,导致gp120发生剧烈的构象变化,从而暴露并激活gp41的膜融合功能。gp41在结构上分为膜外区、跨膜区(tm)和膜内区三部分。其膜外区又包括n末端疏水性融合肽(fp)、n-端螺旋重复序列(nhr)、c-端螺旋重复序列(chr)、近膜区(mper)等几个重要的功能区(图1)。早在1997年,通过解析来源于nhr的多肽n36与来源于chr的多肽c34复合物的晶体结构,发现了典型的六股α-螺旋束结构(6-hb),其中三个nhr通过在a和d位置的氨基酸相互作用形成位于中心的螺旋三聚体,其e和g位置的氨基酸则暴露于中心螺旋体的外围,并与三个chr螺旋在a和d位置的氨基酸相互作用[4]。三个chr螺旋以反向平行的方式分别结合在三个nhr螺旋形成的沟槽中,类似于三个叠在一起的发卡结构。基于6-hb结构对hiv膜融合的机制有了更为深入的理解,首先是暴露的gp41融合肽插入靶细胞膜内,紧接着chr与nhr发生反向结合,通过形成稳定的6-hb将病毒膜与靶细胞膜拉近并导致融合,最终hiv遗传物质进入靶细胞内。6-hb结构还揭示,nhr螺旋的c端部分形成明显的疏水深口袋结构(pocket),而chr的n端即所谓口袋结合区(pbd)的三个氨基酸插入到nhr疏水口袋,其间的相互作用对稳定6-hb结构起着重要作用,因此也是hiv感染所必需。长期以来,nhr疏水口袋结构被认为是抗hiv药物的重要靶点,而chr的pbd基序则是设计抗hiv多肽抑制剂的关键所在[5,6]。
已有的研究表明,来源于gp41nhr和chr的多肽具有显著的抗hiv活性,主要是通过与对应的nhr或chr结合而竞争性地阻断病毒本身6-hb的形成,从而阻断病毒-细胞膜融合过程[6]。通常原型chr多肽的抗病毒活性要比原型nhr多肽显著为高。药物t-20属于chr多肽,其序列见图1所示,对应于hiv-1毒株hxb2gp41的第127位至162位氨基酸序列。t-20在序列结构上的一个特点是其c末端为一富含色氨酸的疏水基序(trm:waslwnwf),但其n末端不包括pbd序列(wmewdrei)。研究发现,t-20的trm具有介导多肽与细胞脂膜结合的作用,所以被认为是脂膜结合区(lbd),这种特性对t-20的抗病毒活性非常重要。随着t-20在临床应用中所表现的明显缺陷,研发新一代hiv膜融合抑制剂一直是国际热点课题,但大都是基于含有34个氨基酸的chr多肽c34作为模板,罕见以t-20作为模板的报道。这可能因为:1)c34被率先用于6-hb结构的解析,其对应的gp41序列第117-150位氨基酸被认为chr核心序列;2)c34的n末端包含重要的pbd序列,比t-20具有较强的nhr结合能力以及抗病毒活性;3)c34对t-20耐药病毒株的抑制活性显著提高。新研发的hiv膜融合抑制剂如t2635、sc35ek、sc29ek、西夫韦肽(sft)、艾博卫泰(abt)、c34-chol等都是通过对c34进行序列优化和/或修饰而获得的[6,7],它们也确实比t-20具有更好的抑制活性以及稳定性等优势。
最近,chr多肽“m-t钩子”结构的发现对设计高活性hiv膜融合抑制剂提供了新的手段[8-10]。研究表明,在chr多肽的pbd前端加入可以形成m-t钩子结构的两个氨基酸(即met115和thr116)可以显著提高抑制剂的靶序列结合能力以及抗病毒活性,特别是提高抑制剂对t-20耐药株的活性,并显著增强抑制剂自身的基因耐药屏障[11,12]。m-t钩子结构也使设计靶向nhr疏水口袋的短肽成为现实,如长度为24个氨基酸的mt-sc22ek,长度为23个氨基酸的hp23和2p23[13-15]。与其他的长序列多肽相比,这些短肽反而具有更高的抗病毒活性以及靶序列结合能力。2p23不但对hiv-1及其t-20耐药性毒株有效,而且对hiv-2及猴免疫缺陷病毒(siv)亦非常有效,是一个广谱的病毒膜融合抑制剂[13]。
细胞膜脂筏结构(lipidraft)富含胆固醇(cholesterol)和鞘磷脂(sphingomyelin)以及许多跨膜蛋白和受体(如hiv受体cd4),对病毒的进入和感染起着重要的作用;另一方面,来源于细胞膜的包膜病毒脂双层膜结构也富含胆固醇和鞘磷脂,参与维持病毒包膜蛋白的正常结构与功能[16,17]。hiv在侵入靶细胞的过程中,脂筏结构以及所含的脂类(如胆固醇和鞘磷脂)给病毒gp120与细胞受体cd4/辅助受体之间的相互作用提供了适宜的平台。研究表明,通过锚定病毒膜融合抑制剂(如多肽、蛋白以及抗体等)到细胞膜表面,能够提高细胞膜局部的抑制剂浓度,从而显著提高其抗病毒活性[18-20]。事实上,基于chr多肽的hiv膜融合抑制剂如t-20、t-1249和西夫韦肽等,多肽自身即具有与细胞膜相互作用的能力[21-23]。通过对t-20的trm氨基酸进行突变分析以及c末端亲脂性功能基团修饰,peisajovich等人揭示了trm对t-20与细胞膜作用而发挥抗病毒功能的重要性[24]。通过重组构建技术将t-20表达于细胞膜表面,也能够显著提高其抑制病毒活性[25,26]。近年的研究也表明,通过对多肽进行脂类化学修饰,即所谓的“脂肽”(lipopeptide),能提高多肽的细胞膜靶向性和抗病毒活性,同时又能显著改善多肽的稳定性和生物半衰期[18-20,27]。针对hiv膜融合抑制剂的研究表明,当chr多肽活性的提高依赖于c末端的修饰,而nhr多肽则适于在n末端修饰,这与6-hb的结构及病毒膜融合的机制相吻合。也就是说,chr多肽通过c末端锚定有益于其与病毒nhr的结合,而nhr多肽则相反,其n末端锚定于细胞膜更有利于其与病毒chr的结合[19,28,29]。同设计非修饰的chr多肽一样,脂肽hiv膜融合抑制剂的设计聚焦于包含pbd的c34作为模板。代表性的例子是2009年ingallinella等人设计的脂肽c34-chol(见图1),它是将胆固醇通过一个柔性连接臂和半胱氨酸连接到c34的c末端,根据其抗病毒结果被认为是目前活性最高的hiv膜融合抑制剂,而且其在动物体内的代谢半衰期也显著延长[20]。本发明人的实验室利用棕榈酸(c16)、胆固醇以及二氢鞘氨醇(dihydrosphingosine)三种脂质化合物分别对靶向nhr口袋的短肽hp23和hp23l进行了修饰,制备了一组高活性脂肽,其中lp-11的体内稳定性也得到大幅度提高[18]。最近,本发明人的实验室又设计了一个基于广谱抗hiv短肽2p23的棕榈酸修饰脂肽lp-19,其具有更高的抗病毒活性和成药性[30]。这些研究进展都为设计新的hiv膜融合抑制剂奠定了坚实的理论基础和技术路线。
参考文献:
1.flexnerc.hivdrugdevelopment:thenext25years.natrevdrugdiscov2007,6:959-966.
2.esteja,telentia.hiventryinhibitors.lancet2007,370:81-88.
3.eckertdm,kimps.mechanismsofviralmembranefusionanditsinhibition.annurevbiochem2001,70:777-810.
4.chandc,fassd,bergerjm,kimps.corestructureofgp41fromthehivenvelopeglycoprotein.cell1997,89:263-273.
5.chandc,chutkowskict,kimps.evidencethataprominentcavityinthecoiledcoilofhivtype1gp41isanattractivedrugtarget.procnatlacadsciusa1998,95:15613-15617.
6.hey.synthesizedpeptideinhibitorsofhiv-1gp41-dependentmembranefusion.currpharmdes2013,19:1800-1809.
7.egginkd,berkhoutb,sandersrw.inhibitionofhiv-1byfusioninhibitors.currpharmdes2010,16:3716-3728.
8.chongh,yaox,sunj,qiuz,zhangm,walterspergers,etal.them-thookstructureiscriticalfordesignofhiv-1fusioninhibitors.jbiolchem2012,287:34558-34568.
9.chongh,qiuz,suy,hey.then-terminalt-tmotifofathird-generationhiv-1fusioninhibitorisnotrequiredforbindingaffinityandantiviralactivity.jmedchem2015,58:6378-6388.
10.chongh,yaox,qiuz,qinb,hanr,walterspergers,etal.discoveryofcriticalresiduesforviralentryandinhibitionthroughstructuralinsightofhiv-1fusioninhibitorcp621-652.jbiolchem2012,287:20281-20289.
11.chongh,yaox,qiuz,sunj,qiaoy,zhangm,etal.them-thookstructureincreasesthepotencyofhiv-1fusioninhibitorsifuvirtideandovercomesdrugresistance.jantimicrobchemother2014,69:6759.
12.chongh,qiuz,sunj,qiaoy,lix,hey.twom-thookresiduesgreatlyimprovetheantiviralactivityandresistanceprofileofthehiv-1fusioninhibitorsc29ek.retrovirology2014,11:40.
13.xiongs,borregop,dingx,zhuy,martinsa,chongh,etal.ahelicalshort-peptidefusioninhibitorwithhighlypotentactivityagainsthumanimmunodeficiencyvirustype1(hiv-1),hiv-2,andsimianimmunodeficiencyvirus.jvirol2017,91:e01839-16.
14.chongh,qiuz,suy,yangl,hey.designofahighlypotenthiv-1fusioninhibitortargetingthegp41pocket.aids2015,29:13-21.
15.chongh,yaox,qiuz,sunj,zhangm,walterspergers,etal.short-peptidefusioninhibitorswithhighpotencyagainstwild-typeandenfuvirtide-resistanthiv-1.fasebj2013,27:1203-1213.
16.bruggerb,glassb,haberkantp,leibrechti,wielandft,krausslichhg.thehivlipidome:araftwithanunusualcomposition.procnatlacadsciusa2006,103:2641-2646.
17.onoa,freedeo.plasmamembraneraftsplayacriticalroleinhiv-1assemblyandrelease.procnatlacadsciusa2001,98:13925-13930.
18.chongh,wux,suy,hey.developmentofpotentandlong-actinghiv-1fusioninhibitors.aids2016,30:1187-1196.
19.wexler-coheny,shaiy.membrane-anchoredhiv-1n-heptadrepeatpeptidesarehighlypotentcellfusioninhibitorsviaanalteredmodeofaction.plospathog2009,5:e1000509.
20.ingallinellap,bianchie,ladwana,wangyj,hrinr,venezianom,etal.additionofacholesterolgrouptoanhiv-1peptidefusioninhibitordramaticallyincreasesitsantiviralpotency.procnatlacadsciusa2009,106:5801-5806.
21.matospm,castanhoma,santosnc.hiv-1fusioninhibitorpeptidesenfuvirtideandt-1249interactwitherythrocyteandlymphocytemembranes.plosone2010,5:e9830.
22.franquelimhg,louralm,santosnc,castanhoma.sifuvirtidescreensrigidmembranesurfaces.establishmentofacorrelationbetweenefficacyandmembranedomainselectivityamonghivfusioninhibitorpeptides.jamchemsoc2008,130:6215-6223.
23.veigaas,santosnc,louralm,fedorova,castanhoma.hivfusioninhibitorpeptidet-1249isabletoinsertoradsorbtolipidicbilayers.putativecorrelationwithimprovedefficiency.jamchemsoc2004,126:14758-14763.
24.peisajovichsg,gallosa,blumenthalr,shaiy.c-terminaloctylationrescuesaninactivet20mutant:implicationsforthemechanismofhiv/simianimmunodeficiencyvirus-inducedmembranefusion.jbiolchem2003,278:21012-21017.
25.hildingerm,dittmarmt,schult-dietrichp,fehseb,schnierlebs,thalers,etal.membrane-anchoredpeptideinhibitshumanimmunodeficiencyvirusentry.jvirol2001,75:3038-3042.
26.egelhoferm,brandenburgg,martiniush,schult-dietrichp,melikyang,kunertr,etal.inhibitionofhumanimmunodeficiencyvirustype1entryincellsexpressinggp41-derivedpeptides.jvirol2004,78:568-575.
27.ashkenazia,viardm,ungerl,blumenthalr,shaiy.sphingopeptides:dihydrosphingosine-basedfusioninhibitorsagainstwild-typeandenfuvirtide-resistanthiv-1.fasebj2012,26:4628-4636.
28.wexler-coheny,shaiy.demonstratingthec-terminalboundaryofthehiv1fusionconformationinadynamicongoingfusionprocessandimplicationforfusioninhibition.fasebj2007,21:3677-3684.
29.wexler-coheny,ashkenazia,viardm,blumenthalr,shaiy.virus-cellandcell-cellfusionmediatedbythehiv-1envelopeglycoproteinisinhibitedbyshortgp41n-terminalmembrane-anchoredpeptideslackingthecriticalpocketdomain.fasebj2010,24:4196-4202.
30.chongh,xuej,xiongs,congz,dingx,zhuy,liuz,chent,fengy,hel,guoy,weiq,zhouy,qinc,hey.alipopeptidehiv-1/2fusioninhibitorwithhighlypotentinvitro,exvivoandinvivoantiviralactivity.jvirol2017,91:e00288-17.
发明公开
本发明所要解决的技术问题是如何强效抑制hiv。
为了解决以上技术问题,本发明提供了强效hiv膜融合抑制剂。本发明所提供的强效hiv膜融合抑制剂,是具有极强抑制hiv活性的脂肽、其药用盐、或其衍生物,其中,所述脂肽为下述a)或b):
a)所述脂肽由具有抗病毒活性的多肽和与所述多肽的羧基末端相连的亲脂性化合物连接而成;
b)所述脂肽由具有抗病毒活性的多肽、末端保护基和与所述多肽的羧基末端相连的亲脂性化合物连接而成;所述末端保护基为氨基端保护基和/或羧基端保护基;
a)和b)中,所述多肽为p1至p5中的任一种:
所述p1的序列为下述(式ⅰ)所示,
(式ⅰ)
x1x2x3x4x5x6x7x8x9x10x11x12x13x14x15x16x17x18x19x20x21x22x23x24x25x26x27x28
(式ⅰ)中,
x1至x28均分别为一个氨基酸残基,所述x1为w、l或y,所述x2为e或t,所述x3为q、a或s,所述x4为k、n或l,x5为i或l,x6为e、d、k、r或a,x7为e、d、k、r或a,x8为l或i,x9为l或i,x10为k、r、e、d或a,x11为k、r、e、d或a,x12为a或s,x13为e、d、k、r或a,x14为e、d、k、r或a,x15为q,x16为q,x17为k、r、e、d或a,x18为k、r、e、d或a,x19为n,x20为e或d,x21为e、d、k、r或a,x22为e、d、k、r或a,x23为l或i,x24为k、r、e、d或a,x25为k、r、e、d或a,x26为l或i,x27为e或d,x28为k或r;
所述p2为缺失所述p1的氨基末端的1至4个氨基酸残基(即式ⅰ中x1、x2、x3和x4这四个氨基酸残基中的1至4个氨基酸残基)得到的多肽;
所述p3为缺失所述p1的羧基末端的1至3个氨基酸残基(即式ⅰ中x26、x27和x28这三个氨基酸残基中的1至3个氨基酸残基)得到的多肽;
所述p4为在所述p1的羧基末端添加半胱氨酸残基得到的多肽;
所述p5的序列为下述(式ⅱ)所示,
(式ⅱ)
x5x6x7x8x9x10x11x12x13x14x15x16x17x18x19x20x21x22x23x24x25
(式ⅱ)中,x5至x25的定义同(式ⅰ);
所述病毒为下述v1-v7中的任一:v1、hiv-1、hiv-2和siv;v2、hiv-1和hiv-2;v3、hiv-1和siv;v4、hiv-2和siv;v5、hiv-1;v6、hiv-2;v7、siv。
实验证明,上述p5为本发明脂肽的核心序列。在该核心序列的n末端加上1至4个氨基酸残基和/或在其c末端加上1-3个氨基酸残基均能有效提高其抗病毒活性。
上述脂肽、其药用盐、或其衍生物中,所述脂肽的抗病毒活性高于lp-19和/或t-20和/或c34-chol。
所述p1的序列为x1x2x3x4ieelx9kkx12eeqqkkneeelkklek;
所述p2为p2-1、p2-2、p2-3或p2-4,
所述p2-1的序列为x2x3x4ieelx9kkx12eeqqkkneeelkklek;
所述p2-2的序列为x3x4ieelx9kkx12eeqqkkneeelkklek;
所述p2-3的序列为x4ieelx9kkx12eeqqkkneeelkklek;
所述p2-4的序列为ieelx9kkx12eeqqkkneeelkklek;
所述p3的序列为x1x2x3x4ieelx9kkx12eeqqkkneeelkk;
所述p4的序列为x1x2x3x4ieelx9kkx12eeqqkkneeelkklekc;
p1、p2-1、p2-2、p2-3、p2-4、p3和p4中,x1、x2、x3、x4、x9和x12的定义同(式ⅰ)。
上述脂肽、其药用盐、或其衍生物中,所述多肽的序列中除xn(n为1-28中的任一个自然数)外,其他每个大写字母均为氨基酸的缩写,氨基酸的缩写具有本领域公知的含义,例如:y为酪氨酸、t为苏氨酸、s为丝氨酸、l为亮氨酸、i为异亮氨酸、e为谷氨酸、k为赖氨酸、q为谷氨酰胺、n为天冬酰胺、a为丙氨酸、w为色氨酸等。所述多肽序列中的所有氨基酸可为l型氨基酸,其中的一个或多个(如2-5个、2-4个或2-3个)氨基酸也可以用构象为d型的氨基酸、人工修饰的氨基酸、自然界存在的稀有氨基酸等进行替换,以提高多肽的生物利用度、稳定性和/或抗病毒活性。其中d型氨基酸是指与组成蛋白质的l型氨基酸相对应的氨基酸;人工修饰的氨基酸指经过甲基化、磷酸化等修饰的组成蛋白质的常见l型氨基酸;自然界存在的稀有氨基酸包括组成蛋白质的不常见氨基酸和不组成蛋白质的氨基酸,例如5-羟基赖氨酸、甲基组氨酸、γ氨基丁酸、高丝氨酸等。
上述脂肽、其药用盐、或其衍生物中,所述p1为p-80/84/85/52、p-87/51或p50;所述p-80/84/85/52为序列表中序列1所示的多肽(即图2中lp-80、lp-84、lp-85和lp-52的第1-28位氨基酸残基所示的多肽),所述p-87/51为序列表中序列2所示的多肽(即图2中lp-87和lp-51的第1-28位氨基酸残基所示的多肽),所述p50为序列表中序列3所示的多肽(即图2中lp-50的第1-28位氨基酸残基所示的多肽);所述p2-1为p-88/62,所述p-88/62为序列表中序列4所示的多肽(即图2中lp-88和lp-62的第1-27位氨基酸残基所示的多肽);所述p2-2为p63或p60,所述p63为序列表中序列5所示的多肽(即图2中lp-63的第1-26位氨基酸残基所示的多肽),所述p60为序列表中序列6所示的多肽(即图2中lp-60的第1-26位氨基酸残基所示的多肽);所述p2-3为p-89/64,所述p-89/64为序列表中序列7所示的多肽(即图2中lp-89和lp-64的第1-25位氨基酸残基所示的多肽);所述p2-4为p-90/65或p61,所述p-90/65为序列表中序列8所示的多肽(即图2中lp-90和lp-65的第1-24位氨基酸残基所示的多肽),所述p61为序列表中序列9所示的多肽(即图2中lp-61的第1-24位氨基酸残基所示的多肽);所述p3为p-91/55,所述p-91/55为序列表中序列10所示的多肽(即图2中lp-91和lp-55的第1-25位氨基酸残基所示的多肽);所述p4为p83或p86,所述p83为序列表中序列11所示的多肽(即图2中lp-83的第1-29位氨基酸残基所示的多肽),所述p86为序列表中序列12所示的多肽(即图2中lp-86的第1-29位氨基酸残基所示的多肽)。
上述脂肽、其药用盐、或其衍生物中,所述亲脂性化合物可为含8到20个碳原子的脂肪酸(fattyacid)、胆固醇(cholesterol,chol)、二氢(神经)鞘氨醇(dihydrosphingosine,dhs)或维生素e(tocopherol,toc)等。
上述脂肽、其药用盐、或其衍生物中,所述含8到20个碳原子的脂肪酸可为棕榈酸(又名软脂酸)(c16)或硬脂酸(c18)。
上述脂肽、其药用盐、或其衍生物中,亲脂性化合物可以连接在末端氨基酸的侧链上,也可直接连接在肽链上。其中,c末端连接的亲脂性化合物中的脂肪酸、二氢鞘氨醇和维生素e的修饰可通过与多肽末端的赖氨酸(lys)侧链的氨基进行酰胺化反应而完成;胆固醇的修饰可通过与多肽末端的半胱氨酸(cys)侧链的巯基与溴乙酸胆固醇酯进行化学选择性极高的成硫醚反应接枝到多肽链上。
上述脂肽、其药用盐、或其衍生物中,所述脂肽可为lp-80/84/85/52、lp-90/65、lp-87/51、lp-88/62、lp-50、lp-83、lp-91/55、lp-86、lp-63、lp-89/64、lp-60和lp-61这12种脂肽中的任一种;
所述lp-80/84/85/52为lp-80/84/85/52a或lp-80/84/85/52b;所述lp-80/84/85/52a由所述p-80/84/85/52和与所述p-80/84/85/52的羧基末端相连的亲脂性化合物连接而成;所述lp-80/84/85/52b由所述lp-80/84/85/52a与所述末端保护基连接而成;所述lp-80/84/85/52a和lp-80/84/85/52b中,所述亲脂性化合物为硬脂酸、二氢鞘氨醇、维生素e或棕榈酸;
所述lp-90/65为lp-90/65a或lp-90/65b,所述lp-90/65a由所述p-90/65和与所述p-90/65的羧基末端相连的亲脂性化合物连接而成;所述lp-90/65b由所述lp-90/65a与所述末端保护基连接而成;所述lp-90/65a和lp-90/65b中,所述亲脂性化合物为硬脂酸或棕榈酸;
所述lp-87/51为lp-87/51a或lp-87/51b;所述lp-87/51a由所述p-87/51和与所述p-87/51的羧基末端相连的亲脂性化合物连接而成;所述lp-87/51b由所述lp-87/51a与所述末端保护基连接而成;所述lp-87/51a和lp-87/51b中,所述亲脂性化合物为二氢鞘氨醇或棕榈酸;
所述lp-88/62为lp-88/62a或lp-88/62b;所述lp-88/62a由所述p-88/62和与所述p-88/62的羧基末端相连的亲脂性化合物连接而成;所述lp-88/62b由所述lp-88/62a与所述末端保护基连接而成;所述lp-88/62a和lp-88/62b中,所述亲脂性化合物为硬脂酸或棕榈酸;
所述lp-50为lp-50a或lp-50b;所述lp-50a由所述p50和与所述p50的羧基末端相连的棕榈酸连接而成;所述lp-50b由所述lp-50a与所述末端保护基连接而成;
所述lp-83为lp-83a或lp-83b;所述lp-83a由所述p83和与所述p83的羧基末端相连的胆固醇连接而成;所述lp-83b由所述lp-83a与所述末端保护基连接而成;
所述lp-91/55为lp-91/55a或lp-91/55b,所述lp-91/55a由所述p-91/55和与所述p-91/55的羧基末端相连的亲脂性化合物连接而成;所述lp-91/55b由所述lp-91/55a与所述末端保护基连接而成;所述lp-91/55a和lp-91/55b中,所述亲脂性化合物为硬脂酸或棕榈酸;
所述lp-86为lp-86a或lp-86b;所述lp-86a由所述p86和与所述p86的羧基末端相连的胆固醇连接而成;所述lp-86b由所述lp-86a与所述末端保护基连接而成;所述lp-63为lp-63a或lp-63b;所述lp-63a由所述p63和与所述p63的羧基末端相连的棕榈酸连接而成;所述lp-63b由所述lp-63a与所述末端保护基连接而成;所述lp-89/64为lp-89/64a或lp-89/64b;所述lp-89/64a由所述p-89/64和与所述p-89/64的羧基末端相连的亲脂性化合物连接而成;所述lp-89/64b由所述lp-89/64a与所述末端保护基连接而成;所述lp-89/64a和lp-89/64b中,所述亲脂性化合物为硬脂酸或棕榈酸;
所述lp-60为lp-60a或lp-60b;所述lp-60a由所述p60和与所述p60的羧基末端相连的棕榈酸连接而成;所述lp-60b由所述lp-60a与所述末端保护基连接而成;所述lp-61为lp-61a或lp-61b;所述lp-61a由所述p61和与所述p61的羧基末端相连的棕榈酸连接而成;所述lp-61b由所述lp-61a与所述末端保护基连接而成。
上述脂肽、其药用盐、或其衍生物中,本发明的脂肽的氨基端可含有氨基端保护基,所述氨基端保护基可为乙酰基、氨基、马来酰基、琥珀酰基、叔丁氧羰基或苄氧或其他疏水基团或大分子载体基团中的任一基团;本发明的脂肽的羧基端可含有羧基端保护基,所述羧基端保护基可为氨基、酰胺基、羧基、或叔丁氧羰基或其他疏水基团或大分子载体基团中的任一基团。
上述p1-p4中的任一种的多肽、其药用盐、或其衍生物也属于本发明的保护范围。
所述多肽的衍生物具体可为下述1)-5)中的至少一种:
1)所述多肽的氨基端连接氨基端保护基和/或所述多肽的羧基端连接羧基端保护基得到的连接物;
2)所述多肽的羧基端连接寡肽或亲脂性化合物得到的连接物;
3)所述多肽的氨基端连接寡肽或亲脂性化合物得到的连接物;
4)所述多肽的氨基端和羧基端均连接寡肽或亲脂性化合物得到的连接物;
5)所述多肽被蛋白质、聚乙二醇、马来酰亚胺修饰得到的修饰物。
pm1或pm2的多聚体也属于本发明的保护范围:
pm1、由所述脂肽、其药用盐、或其衍生物形成的多聚体;
pm2、由所述多肽、其药用盐、或其衍生物形成的多聚体。
下述组合物也属于本发明的保护范围:一种组合物,其包含c1)和c2):c1)为c11)、c12)或/和c13);所述c11)为所述脂肽、其衍生物、或其可药用盐;所述c12)为所述多肽、其衍生物、或其可药用盐;所述c13)为所述多聚体;
c2)药学上可接受的载体或辅料;
所述组合物具有下述f1)-f5)中的至少一种功能:
f1)抗病毒;
f2)治疗和/或预防和/或辅助治疗病毒感染所致疾病;
f3)抑制病毒进行细胞融合;
f4)抑制病毒侵入细胞;
f5)抑制病毒复制;
f1)-f5)中,所述病毒为下述v1-v7中的任一:
v1、hiv-1、hiv-2和siv;
v2、hiv-1和hiv-2;
v3、hiv-1和siv;
v4、hiv-2和siv;
v5、hiv-1;
v6、hiv-2;
v7、siv。
上述c11)、c12)、c13)或/和c14)在制备e1)-e5)中至少一种产品中的应用也属于本发明的保护范围:
所述c14)为所述组合物;
所述e1)为抗病毒的产品,如药物或疫苗;
所述e2)为治疗和/或预防和/或辅助治疗病毒感染所致疾病(如艾滋病)的产品,如药物或疫苗;
所述e3)为抑制病毒进行细胞融合的产品,如药物或疫苗;
所述e4)为抑制病毒侵入细胞的产品,如药物或疫苗;
所述e5)为抑制病毒复制的产品,如药物或疫苗;
所述e1)-e5)中,所述病毒为下述v1-v7中的任一:
v1、hiv-1、hiv-2和siv;
v2、hiv-1和hiv-2;
v3、hiv-1和siv;
v4、hiv-2和siv;
v5、hiv-1;
v6、hiv-2;
v7、siv。
本发明的脂肽药用盐和多肽药用盐,包括醋酸盐(acetate)、乳糖醛酸盐(lactobionate)、苯磺酸盐(benzenesulfonate)、月桂酸酯(laurate)、安息香酸盐(benzoate)、苹果酸盐(malate)、重碳酸盐(bicarbonate)、马来酸盐(maleate)、硫酸氢盐(bisulfate)、扁桃酸盐(mandelate)、酒石酸氢盐(bitartrate),甲磺酸盐(mesylate),硼酸盐(borate),溴甲烷(methylbromide),溴化物(bromide),硝酸甲酯(methylnitrate),依地酸钙(calciumedetate),甲基硫酸盐(methylsulfate),右旋樟脑磺酸(camsylate),粘酸盐(mucate),碳酸盐(carbonate),萘磺酸盐(napsylate),氯化物(chloride),硝酸盐(nitrate),棒酸盐(clavulanate),n-甲葡糖胺(n-methylglucamine),柠檬酸盐(citrate),铵盐(ammoniumsalt),二氢氯化物(dihydrochloride),油酸盐(oleate),乙二胺四乙酸盐(edetate),草酸盐(oxalate),乙二磺酸盐(edisylate),扑酸盐(pamoate)(双羟萘酸盐embonate),丙酸酯月桂硫酸酯(estolate),棕榈酸盐(palmitate),乙磺酸酯(esylate),泛酸盐(pantothenate),延胡索酸盐(fumarate),磷酸盐/二磷酸(phosphate/diphosphate),葡庚糖酸盐(gluceptate),聚半乳糖醛酸盐(polygalacturonate),葡(萄)糖酸盐(gluconate),水杨酸盐(salicylate),谷氨酸盐(glutamate),硬脂酸盐(stearate),对羟乙酰氨基苯胂酸(glycollylarsanilate),硫酸盐(sulfate),羟基苯甲酸盐(hexylresorcinate),碱式乙酸盐(subacetate),海巴(hydrabamine),琥珀酸盐(succinate),氢溴酸盐(hydrobromide),丹宁酸盐(tannate),氢氯化物(hydrochloride),酒石酸盐(tartrate),羟萘酸盐(hydroxynaphthoate),8-氯茶碱盐(teoclate),碘化物(iodide),甲苯磺酸盐(tosylate),三乙基碘(triethiodide),乳酸(lactate),戊酸盐(valerate)等。取决于用途,药用盐可以由阳离子如钠(sodium)、钾(potassium)、铝(aluminum)、钙(calcium)、锂(lithium)、锰(magnesium)和锌(zinc)、铋(bismuth)等所形成,也可由碱如氨、乙二胺(ethylenediamine)、n-甲基-谷氨酰胺(n-methyl-glutamine)、赖氨酸(lysine)、精氨酸(arginine)、鸟氨酸(ornithine)、胆碱(choline)、n,n'-二苄基乙二胺(n,n'-dibenzylethylene-diamine),氯普鲁卡因(chloroprocaine),二乙醇氨(diethanolamine),普鲁卡因(procaine),二乙胺(diethylamine),哌嗪(piperazine),三羟甲基氨基甲烷(tris(hydroxymethyl)aminomethane)和羟化四甲铵(tetramethylammoniumhydroxide)等所形成。这些盐可以采用标准方法制备,例如通过游离酸与有机或无机碱的反应。在一个碱性基团如氨基存在的情况下,酸性盐如氢氯化物(hydrochloride)、氢溴化物(hydrobromide)、醋酸盐(acetate)、扑酸盐(pamoate)等等可用作剂型;在一个酸性基团(如-cooh)或醇基存在的情况下,可药用的酯如醋酸酯(acetate)、马来酸酯(maleate)、三甲基乙酸氯甲酯(pivaloyloxymethyl)等、以及文献中公知的用于改善可溶性和水解性的酯可以用作持续释放和前体药制剂。
本发明中,所述抗病毒活性也可称为抑制病毒活性,具体可为抑制病毒进行细胞融合和/或抑制病毒侵入细胞和/或抑制病毒复制。在非人灵长类动物(猴子)体内表现出显著的长效抗病毒作用。
本发明所提供的脂肽或多肽、其衍生物、或其药用盐,所述多聚体或所述组合物,可以用于hiv(hiv-1和/或hiv-2)和/或siv感染的治疗,包括hiv和/或siv感染的各个阶段,例如艾滋病发病期(aids)、有症状期和无症状期。本发明所提供的脂肽或多肽、其衍生物、或其药用盐,所述多聚体或所述组合物,也可以用于hiv(hiv-1和/或hiv-2)和/或siv感染的预防,包括暴露前或可疑暴露后,例如输血、器官移植、体液交换、咬伤、意外针刺或手术中暴露于病人血液等。
在实际应用中,可以将本发明的脂肽或多肽、其衍生物、或其可药用盐,所述多聚体或所述组合物作为药物直接给予病人、或者与适宜的载体或赋形剂混合后给予病人,以达到治疗和/或预防hiv感染的目的。这里的载体材料包括但不限于水溶性载体材料(如聚乙二醇、聚乙烯吡咯烷酮、有机酸等)、难溶性载体材料(如乙基纤维素、胆固醇硬脂酸酯等)、肠溶性载体材料(如醋酸纤维素酞酸酯和羧甲乙纤维素等)。其中优选的是水溶性载体材料。使用这些材料可以制成多种剂型,包括但不限于片剂、胶囊、滴丸、气雾剂、丸剂、粉剂、溶液剂、混悬剂、乳剂、颗粒剂、脂质体、透皮剂、口含片、栓剂、冻干粉针剂等。其中,栓剂可为阴道栓剂,也可以是阴道环,也可以是适于阴道应用的药膏、乳霜或凝胶。可以是普通制剂、缓释制剂、控释制剂及各种微粒给药系统。为了将单位给药剂型制成片剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如稀释剂与吸收剂,如淀粉、糊精、硫酸钙、乳糖、甘露醇、蔗糖、氯化钠、葡萄糖、尿素、碳酸钙、白陶土、微晶纤维素、硅酸铝等;湿润剂与粘合剂,如水、甘油、聚乙二醇、乙醇、丙醇、淀粉浆、糊精、糖浆、蜂蜜、葡萄糖溶液、阿拉伯胶浆、明胶浆、羧甲基纤维素钠、紫胶、甲基纤维素、磷酸钾、聚乙烯吡咯烷酮等;崩解剂,例如干燥淀粉、海藻酸盐、琼脂粉、褐藻淀粉、碳酸氢钠与枸橼酸、碳酸钙、聚氧乙烯、山梨糖醇脂肪酸酯、十二烷基磺酸钠、甲基纤维素、乙基纤维素等;崩解抑制剂,例如蔗糖、三硬脂酸甘油酯、可可脂、氢化油等;吸收促进剂,例如季铵盐、十二烷基硫酸钠等;润滑剂,例如滑石粉、二氧化硅、玉米淀粉、硬脂酸盐、硼酸、液体石蜡、聚乙二醇等。还可以将片剂进一步制成包衣片,例如糖包衣片、薄膜包衣片、肠溶包衣片,或双层片和多层片。为了将单位给药剂型制成丸剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如稀释剂与吸收剂,如葡萄糖、乳糖、淀粉、可可脂、氢化植物油、聚乙烯吡咯烷酮、gelucire、高岭土、滑石粉等;粘合剂如阿拉伯胶、黄蓍胶、明胶、乙醇、蜂蜜、液糖、米糊或面糊等;崩解剂,如琼脂粉、干燥淀粉、海藻酸盐、十二烷基磺酸钠、甲基纤维素、乙基纤维素等。为了将单位给药剂型制成栓剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如聚乙二醇、卵磷脂、可可脂、高级醇、高级醇的酯、明胶、半合成甘油酯等。为了将单位给药剂型制成注射用制剂,如溶液剂、乳剂、冻干粉针剂和混悬剂,可以使用本领域常用的所有稀释剂,例如,水、乙醇、聚乙二醇、1,3-丙二醇、乙氧基化的异硬脂醇、多氧化的异硬脂醇、聚氧乙烯山梨醇脂肪酸酯等。另外,为了制备等渗注射液,可以向注射用制剂中添加适量的氯化钠、葡萄糖或甘油,此外,还可以添加常规的助溶剂、缓冲剂、ph调节剂等。此外,如需要,也可以向药物制剂中添加着色剂、防腐剂、香料、矫味剂、甜味剂或其它材料。
使用上述剂型可以经注射给药,包括皮下注射、静脉注射、肌肉注射和腹腔注射、脑池内注射或灌输等;腔道给药,如经直肠、阴道和舌下;呼吸道给药,如经鼻腔;粘膜给药。上述给药途径优选的是注射给药,优选的注射途径是皮下注射。
本发明的脂肽或多肽、其衍生物、或其可药用盐,所述多聚体或所述组合物的给药剂量取决于许多因素,例如所要预防或治疗疾病的性质和严重程度,患者或动物的性别、年龄、体重及个体反应,所用的具体活性成分,给药途径及给药次数等。上述剂量可以单一剂量形式或分成几个,例如二、三或四个剂量形式给药。
对于任何具体的患者,具体的治疗有效剂量水平须根据多种因素而定,所述因素包括所治疗的障碍和该障碍的严重程度;所采用的具体活性成分的活性;所采用的具体组合物;患者的年龄、体重、一般健康状况、性别和饮食;所采用的具体活性成分的给药时间、给药途径和排泄率;治疗持续时间;与所采用的具体活性成分组合使用或同时使用的药物;及医疗领域公知的类似因素。例如,本领域的做法是,活性成分的剂量从低于为得到所需治疗效果而要求的水平开始,逐渐增加剂量,直到得到所需的效果。一般说来,本发明的脂肽、其衍生物、或其可药用盐,所述多聚体或所述组合物用于哺乳动物特别是人的剂量可以介于0.001-1000mg/kg体重/天,例如介于0.01-100mg/kg体重/天,又例如介于0.1-10mg/kg体重/天。给药频率可以为每天1-2次、1次/2天、1次/3天、1次/4天、1次/5天、1次/6天或1次/7天,优选的可以是1次/1-2天或1-2次/周。
本发明的脂肽或多肽、其衍生物、或其可药用盐,所述多聚体或所述组合物可以直接单独用于hiv感染者的治疗和预防,也可以与一种或多种抗hiv药物联合使用,可以同时使用,也可以间隔使用,以达到提高整体治疗效果的目的。这些抗hiv药物包括但不限于逆转录酶抑制剂、蛋白酶抑制剂、侵入抑制剂、整合抑制剂和成熟抑制剂等。上述的逆转录酶抑制剂可以是核苷类逆转录酶抑制剂,如齐多夫定(azt)、拉米夫定(3tc)、去羟肌苷(ddi)、扎西他滨(ddc)、司他夫定(d4t)、替诺福韦(tdf)、阿巴卡韦(abc)、恩曲他滨(ftc),也可以是非核苷类逆转录酶抑制剂,如奈韦拉平(nvp)、依非韦伦(efv)、地拉夫定(dlv)、依曲韦林(etr)等的一种或几种;上述的蛋白酶抑制剂可以是沙奎那韦(sqv-hgc)、茚地那韦(idv)、利托那韦(rtv)、安瑞那韦(apv)、克力芝(lpv/rtv)、奈非那韦(nfv)、福沙那伟钙(fpv)、reyataz(atv)和prezista等的一种或几种;上述的整合抑制剂可以是raltegravir、dolutegravir和elvitegravi等的一种或几种;上述的侵入抑制剂可以是maraviroc、t-20、tak-779、t2635、virip(vir-576)、西夫韦肽、艾博韦肽、可溶性cd4蛋白及其类似物、针对辅助受体ccr5的抗体(如pro140)、针对gp120/gp41的单克隆抗体(如vrc01和10e8)和针对受体cd4的单克隆抗体(如tnx-355)等的一种或几种。
本发明脂肽的设计策略是通过以亲脂性化合物,如长链脂肪酸棕榈酸和硬脂酸、胆固醇、二氢鞘氨醇或维生素e,取代t-20多肽c末端的8个氨基酸的trm序列(waslwnwf),导致的脂肽含有多肽序列对应于t-20的前28个氨基酸,即对应于hiv-1hxb2毒株gp41的第127位至154位氨基酸;进一步通过突变在多肽序列的非nhr结合面氨基酸即b,c和f,g位置对应氨基酸导入有助于形成离子对的ee**kk氨基酸残基,在多肽序列的nhr结合面氨基酸即a和d位置的对应氨基酸导入hiv-2和/或siv的对应氨基酸残基。进一步通过对所产生的脂肽的c端序列和/或n端序列截短,产生一组多肽序列少于28个氨基酸即含有24至27个氨基酸的脂肽,并确定对应于t-20第5位至25位氨基酸即对应于hxb2毒株gp41的第131位至151位氨基酸的序列为本发明强效hiv抑制剂的核心序列(即p5序列)。本发明的多肽具有突出的序列结构特征,在c端连接有亲脂性化合物进行化学修饰,具有显著增强的靶序列结合能力和极强的hiv(hiv-1和/或hiv-2)和/或siv抑制活性,对hiv包膜蛋白(env)介导的细胞融合、病毒进入和感染均具有极强的抑制能力。与t-20相比,本发明的脂肽的抗hiv活性高于数千倍甚至上万倍,也显著高于抗hiv脂肽c34-chol、lp-11和lp-19等具有较高活性的脂肽;同时,它们还具有稳定长效、易于合成、成本低廉等诸多优势。本发明的脂肽对hiv-1各种亚型(如a、b、c、a/e和b/c亚型)、t-20耐药毒株、hiv-2毒株以及猴免疫缺陷病毒(siv)均有极强的抑制活性。
本发明所提供的强效脂肽、其衍生物、或其可药用盐,所述多聚体或所述组合物,可以用于hiv(hiv-1和/或hiv-2)和/或siv感染的治疗和/或预防。在实际应用中,可以将本发明的脂肽、其衍生物、或其可药用盐,所述多聚体或所述组合物作为药物直接给予病人、或者与适宜的载体或赋形剂混合后给予病人,以达到治疗和/或预防hiv感染的目的。
附图说明
图1为hiv融合蛋白gp41的结构与功能及多肽膜融合抑制剂。其中,fp指gp41融合肽;nhr指n-端重复序列;chr指c-端重复序列;tm指跨膜区。箭头所指为“m-t钩子”位置或色氨酸富含基序(trm)位置。gp41示意图上方为nhr多肽n36和n39序列,并分别标注t-20耐药突变基序(t20-resistantsite)和疏水口袋形成基序(pocket-formingsite);下方为chr和基于chr序列的抑制剂序列,其中m-t和pbd序列用下划线表示,trm序列用斜体表示,而本发明多肽序列的突变氨基酸用黑体表示。图中所有多肽或脂肽的氨基末端均乙酰化修饰(ac-)、羧基末端均酰胺化修饰(-nh2)。
图2为hiv膜融合抑制剂的序列结构及其抗病毒活性。其中,t-20的trm序列用斜体表示,m-t钩子和pbd序列用下划线表示。多肽连接臂中,ahx指6-氨基己酸;aeea指8-氨基-3,6-二氧杂辛酸;peg4、peg8和peg12指不同长度聚合的聚乙二醇,其中,peg4为fmoc-nh-peg4-ch2ch2cooh,peg8为fmoc-nh-peg8-ch2ch2cooh,peg12为fmoc-nh-peg12-ch2ch2cooh。c16代表棕榈酸,c18代表硬脂酸,chol代表胆固醇,dhs代表二氢鞘氨醇,toc代表维生素e,c12代表十二烷酸(月桂酸),c8代表辛酸(羊脂酸)。nl4-3假病毒为gp41d36g突变体。实验重复三次,计算平均ic50值。部分强效脂肽用黑体标注。hxb2细胞融合表示hiv-1介导的细胞融合抑制实验结果,nl4-3进入表示hiv-1假病毒介导的细胞进入抑制实验结果,jrcsf复制表示hiv-1复制抑制实验结果。
图3为hiv膜融合抑制剂对各种亚型hiv-1毒株的抑制作用。实验重复三次,计算平均ic50值。
图4为hiv膜融合抑制剂对t-20耐药突变毒株及hiv-2、siv毒株的抑制作用。t-20耐药突变毒株和siv毒株为假病毒,hiv-2为感染性rod毒株。实验重复三次,计算平均ic50值。
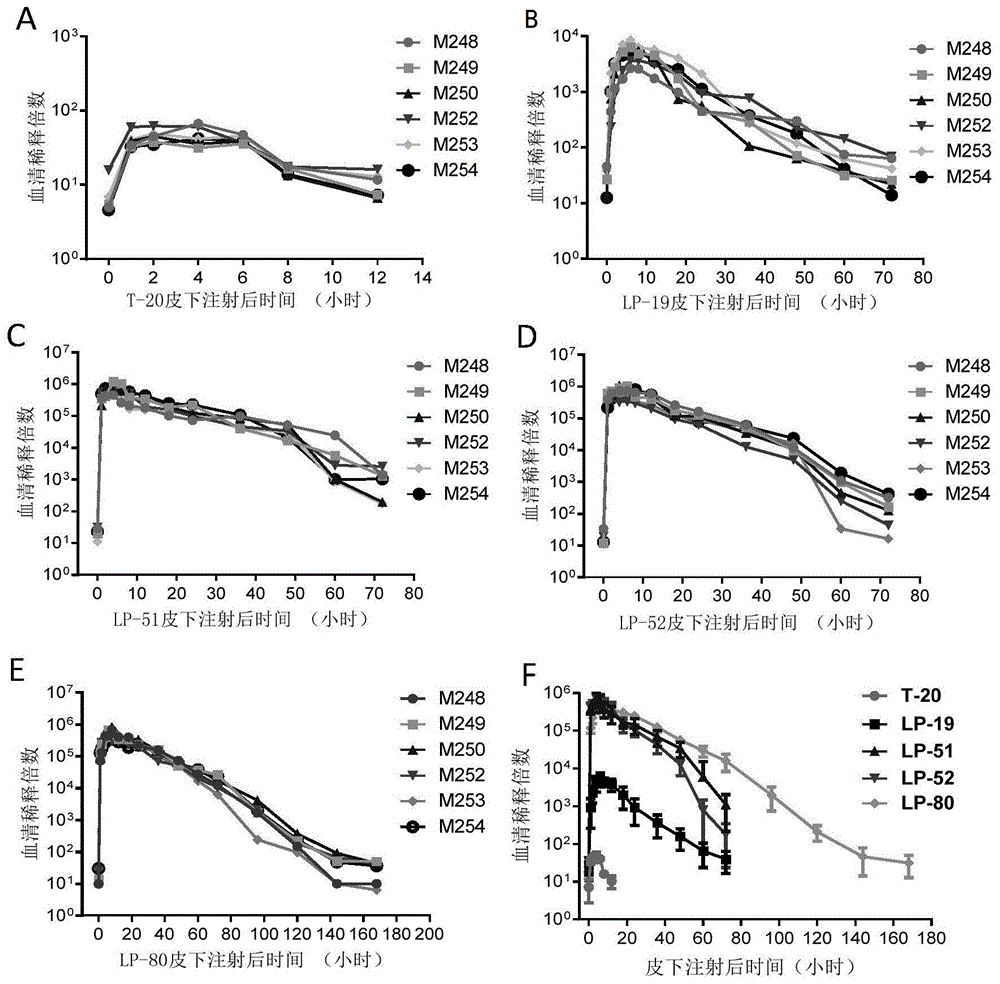
图5为hiv膜融合抑制剂注射猕猴血清的抗病毒活性。图中,m248、m249、m250、m252、m253和m254为猴子的编号。图5中a为t-20注射猕猴血清;图5中b为lp-19注射猕猴血清;图5中c为lp-51注射猕猴血清;图5中d为lp-52注射猕猴血清;图5中e为lp-80注射猕猴血清;图5中f为血清抗病毒活性的比较。
图6为hiv膜融合抑制剂与nhr相互作用的圆二色谱分析。抑制剂的序列结构与本发明的图2相同,其中本发明所述强效脂肽用黑体标注。抑制剂和n39多肽溶于ph为7.2的磷酸盐缓冲液(pbs),终浓度分别为10μm。
图7为t-20和代表性脂肽与nhr相互作用的圆二色谱分析。图7中a为cd扫描结果;图7中b为温度扫描结果。
图8为t-20和代表性脂肽自身二级结构分析。图8中a和图8中b分别为抑制剂在10μ浓度下的cd扫描和温度扫描结果;图8中c和图8中d分别为抑制剂在20μm浓度下的cd扫描和温度扫描结果图;8中e和图8中f分别为抑制剂在40μm浓度下的cd扫描和温度扫描结果。
图9为lp-80在大鼠中的药代动力学。图9中a为给药后lp-80的血清药物浓度检测;图9中b为lp-80的代谢动力学参数。t1/2为末端消除半衰期;cmax为峰浓度;tmax为达峰时间;auc(0-216h)为曲线下面积(0-216h);vz为表观分布容积;cl为清除率;mrt为平均滞留时间;fabs为绝对生物利用度。
图10为lp-80在猴子感染模型的治疗效果。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。下述实施例中的所有多肽的氨基酸均为l型氨基酸。
实施例1.脂肽的制备
本实施例提供的脂肽的结构式为
ac-x1x2x3x4x5x6x7x8x9x10x11x12x13x14x15x16x17x18x19x20x21x22x23x24x25x26x27x28z-nh2,其中x1-x28代表多肽序列,对应于hiv-1病毒株hxb2gp41序列的第127位至154位氨基酸(ytslihslieesqnqqekneqelleldk),其中x1对应127位的y,x2对应128位的t,x3对应129位的s,……x28对应154位的k。通过大量突变获得的全新的序列为强效抑制剂的组成部分,代表性多肽有lp-50、lp-51、lp-52、lp-80、lp-83、lp-84、lp-85、lp-86和lp-87等。x1-x28的定义同(式ⅰ),z为亲脂性化合物,ac为乙酰基,nh2为氨基。
本实施例合成了图2所示的脂肽或多肽,每个脂肽或多肽的氨基末端均连接乙酰基作为氨基端保护基,羧基末端均连接氨基作为羧基端保护基。
其中,棕榈酸(棕榈酸修饰的脂肽:lp-40、lp-41、lp-42、lp-43、lp-44、lp-45、lp-50、lp-51、lp-52、lp-53、lp-54、lp-55、lp-56、lp-57、lp-58、lp-59、lp-60、lp-61、lp-62、lp-63、lp-64、lp-65、lp-66、lp-67、lp-68、lp-69、lp-70、lp-71、lp-72、lp-73、lp-74、lp-75、lp-11、lp-19、c34-c16)、硬脂酸(硬脂酸修饰的脂肽:lp-80、lp-88、lp-89、lp-90、lp-91、lp-92)、二氢鞘氨醇(二氢鞘氨醇修饰的脂肽:lp-84、lp-87)、维生素e(维生素e修饰的脂肽:lp-85)、十二烷酸(十二烷酸修饰的脂肽:lp-81)和辛酸(辛酸修饰的脂肽:lp-82)对多肽的修饰是通过多肽c末端的赖氨酸(lys)的侧链氨基进行酰胺化反应完成,具体参照背景技术参考文献18和27。下面以lp-52和lp-80为例,阐述上述脂肽的合成方法:
所用的化学试剂,如rinkamidembha树脂,各种fmoc氨基酸、棕榈酰氯(palmitoylchloride)、硬脂酰氯(stearoylchloride)、维生素e琥珀酸酯(vitaminesuccinate)、二氢鞘氨醇(d-erythro-dihydrosphingosine)、n,n′-二琥珀酰亚胺基碳酸酯(n,n′-disuccinimidylcarbonate)、n,n'-二异丙基碳二亚胺(dic)、1-羟基苯并三唑(hobt)、三氟乙酸(tfa)、乙二硫醇(edt)、茚三酮、六氢吡啶(pipe)、苯酚、n,n’-二甲基甲酰胺(dmf)、色谱纯乙腈等均从主要化学试剂供应商购买,使用前未经过进一步的提纯。
多肽的合成:采用手动固相fmoc法,以rinkamidembha树脂(取代常数0.34mmol/g)为起始原料,从c端向n端方向合成。用25%六氢吡啶/dmf(体积比)去除rink树脂上的fmoc保护基,然后用2倍当量fmoc-lys(dde)-oh/hobt/dic与树脂进行接枝引入c端第一个氨基酸残基。而后,再次用25%六氢吡啶/dmf(体积比)去除n端fmoc保护基使n端成为自由氨基。如此反复依次连接各个氨基酸残基。所用原料及用量分别对应为fmoc-glu(otbu)-oh(3eq),fmoc-leu-oh(3eq),fmoc-lys(boc)-oh(3eq),fmoc-lys(boc)-oh(3eq),fmoc-leu-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-asn(trt)-oh(3eq),fmoc-lys(boc)-oh(3eq),fmoc-lys(boc)-oh(3eq),fmoc-gln(trt)-oh(3eq),fmoc-gln(trt)-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-ala-oh(3eq),fmoc-lys(boc)-oh(3eq),fmoc-lys(boc)-oh(3eq),fmoc-leu-oh(3eq),fmoc-leu-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-ile-oh(3eq),fmoc-lys(boc)-oh(3eq),fmoc-gln(trt)-oh(3eq),fmoc-glu(otbu)-oh(3eq),fmoc-trp(boc)-oh(3eq)。最后进行n端乙酰化封端(3倍当量ac2o,6倍当量二异丙基乙胺),完成主链的合成。其中各步骤反应时间如下:去保护8分钟,两次;接枝普通氨基酸60分钟。
以上每步反应后都需用dmf洗涤树脂六次以上,并且都通过kaisertest检测来对反应进行控制,若某个氨基酸缩合反应不完全,重复缩合一次,直至得到所需的目标肽段。
多肽的修饰:用2%水合肼/dmf溶液(体积比)处理树脂,以去除c端lys的侧链dde保护基,而后用3倍当量棕榈酰氯或硬脂酰氯与6倍当量二异丙基乙胺混合后与c端lys的侧链氨基进行酰胺化反应(60分钟),从而实现c端lys残基上的棕榈酰化修饰(lp-52)或硬脂酰化修饰(lp-80)。二氢鞘氨醇对多肽的修饰(lp-84、lp-87)是在去除lys的侧链dde保护基后首先加入n,n′-二琥珀酰亚胺基碳酸酯进行活化,然后加入二氢鞘氨醇,反应进行48小时;维生素e对多肽的修饰(lp-85)采用维生素e琥珀酸酯直接与去保护的lys侧链氨基进行酰胺化反应。
切割及去侧链保护:脂肽合成完毕后,真空干燥树脂。往干燥后的树脂中加入切割试剂(三氟乙酸:1,2-乙二硫醇:苯甲硫醚:苯酚:h2o:三异丙基硅烷=68.5:10:10:5:3.5:1,v/v),切割条件为30℃下切割3小时,将目标多肽从树脂上裂解下来并除去侧链保护基。进行过滤操作,将滤液加入到大量冷的无水乙醚中使多肽沉淀析出,离心。用乙醚洗涤数次后干燥,即得到脂肽粗品。
脂肽的纯化及表征:脂肽粗品的纯化在反相高效液相色谱仪上进行,所用色谱柱柱料为10微米(μm)粒径的反相c18或c4硅胶,孔经为
胆固醇修饰的脂肽(lp-83、lp-86、c34-chol)的合成方法参照背景技术中参考文献18和参考文献20进行。首先根据文献中描述的技术路线合成溴乙酸胆固醇酯,然后通过多肽c末端半胱氨酸(cys)侧链的巯基和溴乙酸胆固醇酯进行化学选择性极高的成硫醚反应接枝到多肽链上,即按常规方法合成多肽粗品后,将其溶解于纯dmso中,加入溶于少量三氟乙酸(thf)中的1倍当量胆固醇溴乙酸酯,再加入纯二异丙基乙胺(diea)调节至碱性。用rp-hplc跟踪反应,一般在1小时内即可完成。脂肽的纯化及表征同上,得到的脂肽纯度均大于95%。
实施例2.强效hiv膜融合抑制剂的鉴定
2.1实验材料与方法
将图2中的每一种脂肽和多肽作为待测物,参照背景技术参考文献18进行细胞融合抑制实验、假病毒抑制实验及病毒复制抑制实验鉴定其抗病毒活性。具体方法如下:
hiv-1介导的细胞融合抑制实验:效应细胞hl2/3细胞和靶细胞tzm-bl细胞均由美国国立卫生研究院(nih)艾滋病试剂和参照物项目提供(目录号分别为1294和8129)。两种细胞均为贴壁细胞,用含有氨苄/链霉素双抗和10%胎牛血清(fbs)的dmem细胞培养液进行培养。首先将tzm-bl加入到96孔细胞培养板中(1×104个/孔),于37℃、5%co2的条件下培养过夜。将待测物用dmem细胞培养液进行3倍倍比稀释,并与hl2/3效应细胞混合(3×104个/孔),然后加入到tzm-bl靶细胞,继续培养6小时使之充分融合。然后采用promega公司的荧光素酶报告基因的试剂盒按说明书测定荧光素酶的活性(相对荧光单位,rlu)。计算每一浓度样品抑制率,利用graphpadprismsoftware2.01软件计算半数有效抑制剂量(ic50值)。hiv-1假病毒介导的细胞进入抑制实验:基本步骤包括(1)hiv-1假病毒的制备:采用细胞转染试剂将表达hiv-1毒株nl4-3包膜蛋白(env)的质粒(将背景技术参考文献14的表2的hiv-1毒株nl4-3的d36g突变体的包膜蛋白(env)编码基因插入载体pcdna3.1(-)得到的重组表达质粒)和hiv-1骨架质粒psg3δenv(由美国nih艾滋病试剂和参照物项目提供,目录号为11051)共转染293t细胞,于37℃、5%co2细胞培养箱中孵育6小时后换液,然后继续孵育48小时。用移液器吸取含有假病毒颗粒的细胞培养上清液,经0.45μm滤器过滤收取上清,加入20%胎牛血清(fbs)后转移至聚丙烯管中,于-80摄氏度保存备用或直接进行病毒滴定;(2)hiv-1假病毒的滴定:将病毒在96孔板中做5倍稀释,设置4个复孔8个梯度,终体积为100微升。将tzm-bl细胞用胰酶消化并计数,用dmem完全培养基将细胞稀释至1×105个/ml,每孔加100微升细胞(含15μg/mldeae-dextran),于37℃、5%co2培养48小时。然后从细胞培养箱中取出96孔板,从上样孔中吸弃上清,加入30微升细胞裂解液,放置10分钟后加入100微升荧光素酶检测试剂。用移液器从每孔中吸出100微升液体,加于对应的96孔白板中,于微孔板光度计读取发光值。用reed-muench法计算病毒滴度;(3)抗病毒活性检测:将待测物用dmso溶解并用细胞培养液按3倍倍比稀释,铺入96孔板中,终体积为50微升,其中用50微升dmem培养基替代待测物作为阴性对照。加入100微升浓度为1×105个/ml的tzm-bl靶细胞(含15μg/mldeae-dextran),然后再加入上述获得的hiv-1假病毒50微升(每孔相当于100tcid50),于37℃、5%co2条件下培养48小时后,利用荧光素酶检测试剂(promega)测定每孔的相对荧光单位(rlu)。计算%抑制率和ic50值。
hiv-1复制抑制实验:编码hiv-1病毒株jrcsf的分子克隆质粒pyk-jrcsf由美国nih艾滋病试剂和参照物项目提供(目录号:2708)。采用转染试剂将pyk-jrcsf转染293t细胞,于37℃、5%co2细胞培养箱中孵育6小时后换液,然后继续培养48小时。用移液器轻轻收集含有jrcsf病毒颗粒的细胞培养上清,经0.45微米滤器过滤取上清,加入20%胎牛血清(fbs)后分装于聚丙烯管中,放置-80℃保存备用或直接进行病毒滴定,方法同上述hiv-1假病毒的制备。为测定抗病毒活性,将待测物用dmso溶解并用细胞培养液按3倍倍比稀释,然后铺入96孔细胞培养板中,终体积为50微升,其中用50微升dmem培养基替代待测物作为阴性对照。加入100微升tzm-bl细胞(105个细胞/ml,含15μg/mldeae-dextran),再加入50微升病毒(100tcid50)。于37℃、5%co2条件下培养48小时后,利用荧光素酶检测试剂(promega)测定每孔的相对荧光单位(rlu)。计算%抑制率和ic50值。
2.2实验结果及分析
2.2.1基于t-20的脂肽(lp-40)具有较强的抗病毒活性
为筛选和鉴定强效hiv膜融合抑制剂,本发明独辟蹊径以不含nhr口袋结合区(pbd)的多肽药物t-20作为设计模板。采用三种抗病毒实验方法评价抑制剂对hiv-1介导的细胞融合、假病毒进入和病毒复制的抑制活性(图2)。首先,通过直接删除t-20的c末端的8个氨基酸,合成了一个不含trm基序的多肽t20-trm,发现其在2000nm高浓度下亦没有明显的抗病毒活性,体现了trm对t-20功能的重要性。进而,以棕榈酸(c16)取代t-20的trm合成了脂肽lp-40。令人惊喜的发现是,lp-40比t-20的抗病毒活性显著提高,其对hxb2介导的细胞融合、nl4-3假病毒进入及jrcsf复制的抑制活性分别是t-20的约59倍、21倍和18倍。说明以亲脂性化合物取代trm能够显著改善多肽的抗病毒活性,可以作为hiv膜融合抑制剂设计的重要策略。
2.2.2加入连接臂显著降低lp-40的抗病毒活性
本发明人先前基于对靶向nhr口袋区的短肽(hp23和2p23)进行修饰设计了一组高活性抗hiv脂肽,发现直接将脂类化合物c16、胆固醇和二氢鞘氨醇连接于多肽c末端将导致多肽抗hiv活性的显著下降,而在多肽序列和修饰物之间导入连接臂则显著提高多肽的活性,且随着连接臂长度的增加其抗病毒活性亦增加(见背景技术参考文献18和30)。最终设计的lp-11和lp-19等高活性脂肽具有较长的peg8连接臂,提示连接臂的加入有益于脂肽克服空间位阻而发挥功能。为进一步提高lp-40的活性,设计并合成了lp-41、lp-42、lp-43、lp-44和lp-45五个脂肽,它们分别加入ahx、aeea、peg4、peg8和peg12连接臂(图2)。令人意外的是,连接臂的加入导致脂肽的活性明显下降,且随着连接臂长度的增加活性下降更加明显,这与基于hp23和2p23的脂肽的结果恰恰相反。这些结果提示基于hp23和2p23的脂肽与基于t-20的脂肽有着不同的作用机制,可能与它们在nhr的结合部位不同有关。
2.2.3加入离子对极其显著地提高lp-40的抗病毒活性
本发明进一步尝试通过导入离子对来促进lp-40的螺旋结构和抗病毒活性。本技术通过突变在多肽序列的非nhr结合面的氨基酸(即b,c和f,g位置)引入有助于形成“盐桥结构”的ee**kk氨基酸残基。从图1可见,lp-40多肽序列中的11个氨基酸被e或k取代,从而在i和i+4位置导入三对ee**kk基序,所合成的脂肽命名为lp-50。通过三个抗病毒实验检测lp-50的抑制活性,结果令人难以置信!如图2所示,lp-50抑制细胞融合、假病毒和复制性病毒的ic50值分别在21pm、7pm和23pm,其分别是t-20活性的1151倍、1345倍和226倍,分别是lp-40活性的20倍、63倍和12倍。因此,离子对的导入有可能通过形成“盐桥结构”而提高脂肽的螺旋结构稳定性,从而极大提高脂肽的抗病毒活性。这被后来的圆二色谱检测所证实(见下面实施例7的实验结果)。
2.2.4加入hiv-2/siv氨基酸残基进一步增强lp-50的活性
为进一步改善lp-50的抗病毒活性,本发明进一步尝试通过在多肽nhr结合面即a和d位置或邻近位置导入来源于hiv-2和/或siv相应的氨基酸残基,因此合成了脂肽lp-51和lp-52。所突变的氨基酸见图1所示,lp-51和lp-52的多肽序列仅保留了10个gp41原有序列,与t-20序列仅有不到28%的一致性。抗病毒实验结果显示,lp-51的活性与lp-50相当,而lp-52对hiv-1毒株hbx2介导细胞融合、nl4-3假病毒和复制性jrcsf病毒的抑制活性又进一步分别提高了大约2倍、2倍和5倍。与t-20相比,lp-52在三个实验系统的抑制hiv活性则分别为t-20活性的1859倍、2353倍和1038倍。因此可以说,lp-50、lp-51和lp-52是极强效的hiv膜融合抑制剂。
2.2.5强效抗hiv脂肽核心序列的鉴定
上述强效hiv抑制剂的多肽序列长度为28个氨基酸。为鉴定其关键的序列以及设计含有更短序列脂肽的可能性,本发明首先基于lp-40合成了c末端截短的脂肽lp-53,基于lp-50合成了c末端截短的脂肽lp-54,结果发现其抗病毒能力明显下降(图2)。进一步以lp-52为模板合成了lp-55和lp-56,其中lp-56是以aeea连接臂取代c末端的三个氨基酸残基(lek)。抗病毒实验发现,虽然lp-55和lp-56抑制hxb2细胞融合的活性基本不变,但其抑制nl4-3和jrcsf感染的活性则有所下降(约2倍)。这些实验结果说明脂肽c末端的三个氨基酸残基(lek)对抗病毒活性发挥着重要的作用。
进一步合成了一组n端截短的脂肽(lp-60至lp-68)。抗病毒实验结果发现,基于lp-50截短的两个脂肽即lp-60和lp-61的活性亦有明显的下降;但令人惊奇的是,基于lp-52截短的脂肽lp-62、lp-63和lp-65的活性变化不大,尤其是仅有24个氨基酸的lp-65,其活性与lp-52相当,而含有25个氨基酸的lp-64的活性则明显下降,尤其是对细胞融合的抑制。研究发现,进一步的n端截短则导致脂肽的活性显著下降(lp-66、lp-67)甚或失去抗病毒能力(lp-68)。通过在lp-65的基础上进行截掉c末端lek合成了lp-69,虽然其抗病毒活性显著下降,但相对于仅有21个氨基酸的脂肽而言仍具有强效的病毒抑制活性。这些研究实例结果说明,由21个氨基酸组成的序列“ieelx9kkx12eeqqkkneeelkk”为本发明强效脂肽的核心序列,其对应于t-20的第5位到25位氨基酸序列,也就是对应于hiv-1hxb2毒株gp41的第131位至151位氨基酸序列(ihslieesqnqqekneqelle)。在该核心序列的n端加上weqk(或lean或ytsl)或在其c末端加上lek均能有效提高其抗病毒活性;如果两端氨基酸基序都保留(如lp-52)脂肽活性则可进一步提高。
结果也显示,lp-57比lp-55的抗病毒活性下降了约15到150倍,显示lp-55末端lkk三个氨基酸的重要性,不宜于进一步截短;lp-66的抗病毒活性比lp-65下降了约54到158倍,说明lp-65第一个氨基酸(ile)非常关键,不宜于进一步截短。同时,lp-65与lp-61两个短脂肽相比仅有一个氨基酸的差别(在其第8位分别为s和a),但其活性则相差5到9倍,说明由s到a的替代对本发明强效脂肽非常重要。
与此同时,为揭示强效抗病毒脂肽的序列结构与功能关系,本发明进一步设计合成了一组n端延长的脂肽(图2中lp-70至lp-75),其中lp-74包含了口袋结合区序列(pbd),lp-75同时包含了pbd和m-t钩子形成序列。但令人意外的是,随着多肽序列向n端的延伸,脂肽的抗病毒活性不升反降,尤其是lp-74和lp-75下降比较明显。
2.2.6强效抗hiv脂肽的衍生物及其抗病毒活性
为揭示强效抗hiv脂肽的序列和结构特异性,本发明继续设计和合成了不同亲脂性化合物修饰的脂肽,包括不同链长的脂肪酸、胆固醇、二氢鞘氨醇、维生素e。抗病毒实验结果如图2所示,硬脂酸(c18)修饰的lp-80对nl4-3进入和jrcsf复制的抑制活性甚至比c16修饰的lp-52更有增加,但十二(烷)酸(c12)修饰的lp-81和辛酸(c8)修饰的lp-82活性则显著下降。这四个脂肽具有相同的多肽序列,但脂肪酸链长决定着脂肽的抑制活性。因此,链长为c18和c16的长链脂肪酸对该多肽序列的修饰更为适宜。抗病毒实验结果也表明,胆固醇(lp-83、lp-86)、二氢鞘氨醇(lp-84、lp-87)、维生素e(lp-85)修饰的脂肽也具有强效抗病毒作用。另外,c18修饰的n端截短脂肽lp-88、lp-89和lp-90也具有强效的抗病毒活性。有意思的,拥有25个氨基酸的lp-89比拥有24个氨基酸的lp-90的活性要低,这个现象类似于c16修饰的lp-64和lp-65,因此n端的赖氨酸(k)对基于核心序列的强效短小脂肽没有必要。对于21个氨基酸的核心序列而言,c16和c18修饰脂肽(lp-69和lp-92)的活性基本相当。
与此同时,本发明实施例检测了几个对照脂肽的抗病毒活性,包括lp-11、lp-19、c34-chol和c34-c16(见图2)。可见,对照脂肽均能有效抑制hiv-1介导的细胞融合、进入和复制,其活性明显高于t-20,但显著低于本发明的一些强效脂肽,如c16修饰的lp-52、lp-55和lp-65以及c18修饰的lp-80、lp-90和lp-91等。
实施例3.强效hiv膜融合抑制剂对hiv-1不同亚型的抑制活性
艾滋病主要由hiv-1引起,由于病毒的变异产生了多种亚型,包括a-d、f-h、j和k亚型等。其中a、b和c亚型是引起世界艾滋病流行的主要病毒,而在中国b/c和a/e重组病毒为主。为进一步评价强效hiv膜融合抑制剂的活性,本发明制备了一组35株hiv-1假病毒,包括国际代表毒株和中国目前流行的hiv-1毒株,其中有a亚型3株、b亚型8株、b’亚型4株、c亚型7株、g亚型1株、a/c重组型1株、a/e重组型5株、和b/c重组型6株。制备假病毒的env表达质粒中,除制备pvo、du156和cap210.2.00.e8的env表达质粒由美国nih艾滋病试剂和参照物项目获得外,其它由中国医学科学院病原生物学研究所何玉先教授实验室保存,参见背景技术参考文献13、14和18及chong等发表的文献(chongh,yaox,zhangc,cail,cuis,wangy,hey.biophysicalpropertyandbroadanti-hivactivityofalbuvirtide,a3-maleimimidopropionicacid-modifiedpeptidefusioninhibitor.plosone,2012;7(3):e32599)。假病毒的制备及抗病毒实验的方法同实施例2的2.1中的方法(hiv-1假病毒介导的细胞进入抑制实验)。为便于比较分析,本实施例测定了t-20、lp-40、lp-50、lp-51、lp-52、lp-55、lp-65、lp-80、lp-85、lp-90以及对照脂肽lp-19、c34-chol共计12个抑制剂对上述35种假病毒的抑制活性。结果见图3所示,t-20、lp-40、lp-50、lp-51、lp-52、lp-55、lp-65、lp-80、lp-85和lp-90抑制各型hiv-1假病毒的平均ic50值分别为41410pm、6369pm、41pm、33pm、16pm、34pm、52pm、6pm、44pm和14pm。可见,本发明新合成脂肽对不同亚型hiv-1的抑制活性比t-20显著提高,依次为t-20的7倍、1010倍、1255倍、2588倍、1218倍、796倍、6902倍、941倍和2958倍。其中,lp-80呈现最强的病毒抑制活性,针对35种假病毒的平均ic50值达6pm,许多毒株的ic50值甚至低于1pm。对照lp-19和c34-chol抑制各型hiv-1假病毒的平均ic50值分别为439pm和66pm,其活性低于lp-50、lp-51、lp-55、lp-65和lp-85,更明显低于lp-52、lp-80和lp-90。通过比较lp-52和lp-80、lp-65和lp-90的ic50值,揭示c18修饰的脂肽的抗病毒活性要优于c16修饰的脂肽。
实施例4.强效hiv膜融合抑制剂对t-20耐药病毒株的抑制活性
t-20是目前唯一批准用于临床的hiv膜融合抑制剂,但其活性不但低于新一代的多肽,而且很容易诱导耐药突变,往往导致临床治疗的失败。为充分体现本发明强效脂肽的抗病毒广谱性和优势,本实施例制备了携带nhr常见t-20耐药突变位点的nl4-3假病毒(如图4,图4中毒株名称的下标即为背景技术参考文献14中表2中的毒株名称)。所用质粒、假病毒制备及抗病毒实验方法参见本发明人发表的文献(背景技术参考文献11、12、14和18)以及上述实施例2和3中的方法。结果见图4所示。首先,与具有代表性的nl4-3d36g突变体相比,野生型nl4-3wt本身即对t-20呈现耐药性,其ic50值分别为13.65nm和152.23nm,但含有单突变或双突变的毒株对t-20的耐药倍数显著升高。结果显示,这些t-20耐药毒株对lp-40的敏感性已有所改善。从实验结果可以发现,在lp-40的基础上导入可以形成离子对盐桥结构所设计的脂肽lp-50对t-20耐药株的抑制活性进一步提高数百甚至近千倍;进一步采用hiv-2/siv序列改造的脂肽lp-51、lp-52、lp-80和lp-85克服耐药株的能力又大大提高,优于lp-40数千到数万倍。通过比较lp-52、lp-55和lp-65、lp-80和lp-90可以发现,c末端截短的脂肽lp-55以及n末端截短的脂肽lp-65和lp-90虽然在前面的抗病毒实验中表现突出,针对大量hiv-1毒株的抑制活性甚至与lp-52和lp-80相差无几,但它们对t-20耐药突变毒株的活性则比lp-52和lp-80差得多。值得特别强调的是,虽然本发明强效抑制剂的代表性脂肽包括lp-50、lp-51、lp-52、lp-55、lp-65、lp-80、lp-85和lp-90对多数t-20耐药毒株的抑制能力也明显下降,但相对来说它们仍具有很强的抗病毒活性,尤其是lp-52和lp-80的活性仍属于本领域罕见。本实施例也从一个侧面揭示了gp41的nhr序列仍为本发明强效脂肽的主要作用靶点。
实施例5.强效hiv膜融合抑制剂对hiv-2和siv的抑制活性
为进一步体现本发明强效脂肽的抗病毒优势,本实施例检测了它们针对hiv-2和siv的抑制活性。抗病毒实验方法参见本发明人发表的论文(背景技术参考文献13和30)。hiv-2毒株rod(hiv-2rod)的分子克隆质粒prod由葡萄牙里斯本大学nunotaveira教授惠赠,表达siv毒株sivpbj(sivpbj)和siv239包膜蛋白的质粒(分别为psivpbj-env和psiv239)由复旦大学徐建青教授惠赠。感染性rod的制备与上述2.1中感染性jrcsf的制备相同,sivpbj和siv239假病毒的制备同上述实施例2和3中的方法相同。结果如图4所示,t-20对hiv-2和siv毒株的抑制活性极弱,而lp-40的活性仅有微弱的改善。值得点赞的是,所检测的强效脂肽包括lp-50、lp-51、lp-52、lp-65、lp-80、lp-85和lp-90对hiv-2和siv均具有极强的抑制活性。因此,本发明的强效脂肽不但对各种亚型hiv-1高度有效,也对t-20耐药毒株、hiv-2以及siv毒株高度有效,具有极强效且广谱的抗病毒效果。通过比较lp-52、lp-55和lp-65可以发现,n端氨基酸weqk截短对抑制hiv-2和siv的活性影响较小,而c端氨基酸lek的截短则显著影响其活性。
实施例6.强效hiv膜融合抑制剂的体内抗病毒活性
近年的研究表明,基于脂肽的hiv膜融合抑制剂不但抗病毒活性得到改善提高,其体内代谢也比较稳定,因此拥有更长的半衰期。为进一步体现本发明强效脂肽的应用价值和成药优势,本实施例重点分析脂肽lp-51、lp-52和lp-80在体内的抗病毒活性,方法参照发明人发表的文献(背景技术参考文献18和30),通过将抑制剂经皮下途径注射到猴子体内,然后采取不同时间点的血液标本并在体外测定其抗病毒活性,不但可以了解抑制剂在体内的活性,也间接反映其在体内的稳定性。除上述三个强效脂肽外,本实施例包括t-20和lp-19两个对照以便于比较分析。具体方法如下:选择6只实验猕猴(恒河猴),雌雄各半,年龄3-4岁、体重3.4-4.7kg。按每公斤体重3毫克皮下注射t-20、lp-19、lp-51、lp-52或lp-80(均用无菌蒸馏水溶解),分别于注射前(0小时),注射后1、2、4、6、8、12、18、24、36、48、60、72小时抽取0.4ml静脉血标本。lp-80除上述采血时间点外,增加注射后96、120、144和168小时四个采血点。按常规方法分离血清。各抑制剂的注射间隔时间在2周以上,以确保没有上次待测物的残留。按上述实施例中基于假病毒的实验方法检测血清抗hiv-1毒株nl4-3(nl4-3d36g)的活性。血清按3倍倍比稀释。实验结果如图5所示,皮下注射t-20在2和4小时表现为抑制峰值,抑制50%nl4-3感染性的血清最大稀释倍数分别为45倍和46倍(图5中a);皮下注射lp-19在6和8小时表现为抑制峰值,血清最大稀释倍数分别为5396倍和4720倍(图5中b)。然而令人震惊的是,皮下注射lp-51和lp-52均在4和6小时表现为抑制峰值,血清最大稀释倍数lp-51分别为700482倍和584381倍,lp-52分别为700802倍和669112倍(图5中c和d);皮下注射lp-80在6和8小时表现为抑制峰值,血清最大稀释倍数分别为491409倍和537206倍(图5中e)。可见,强效脂肽的血清抑制峰值可为t-20血清抑制峰值的11678倍到15235倍,可为lp-19血清抑制峰值的100倍到130倍(以最高值计算)。更加令人振奋的结果是lp-51、lp-52和lp-80三个脂肽在体内的长效作用,它们即使在注射后72小时(3天)也有较高的血清抑制峰值,依次分别是1122倍、182倍和16157倍。特别是lp-80的血清抑制峰值,在注射后96小时(4天)维持在1980倍,在注射后120小时(5天)维持在211倍,在注射后144小时(6天)与t-20在4小时的峰值一样(46倍)(图5中f)。因此,本发明的脂肽不但强效而且长效。
实施例7.强效hiv膜融合抑制剂与nhr靶序列的相互作用
为探讨强效抗hiv脂肽的作用机制,采用圆二色谱技术(cd)测定抑制剂与nhr靶序列之间的相互作用,包括所形成复合物的二级结构(α-螺旋)和螺旋稳定性(tm值)。圆二色谱仪为日产jasco-815,实验方法参照发明人发表的论文(背景技术参考文献18和30)。来源于nhr的靶序列多肽为n39(见图1),其序列为ac-stmgaasmtltvqarqllsgivqqqnnllraieaqqhll-nh2,对应于t-20在nhr上的结合靶位点。将n39和抑制剂分别溶于磷酸盐缓冲液(pbs)中,配制成浓度为20μm的pbs溶液(ph7.2)。将n39和抑制剂按1:1体积比混合(终浓度分别为10μm),置混合样品于37摄氏度(℃)30分钟使之充分反应,然后在圆二色谱仪上测定其复合物的螺旋含量和tm值。仪器扫瞄波长范围为195-260nm,波长间隔为1nm,扫描三次进行平均。根据cd信号判断多肽间的相互作用情况及螺旋含量。然后,将用于cd信号测定的样品转入温度扫描样品池,将cd仪器程序设为温度扫描,检测波长220nm,扫描范围20-98℃,进行程序扫描得到cd信号随温度变化曲线,据此计算tm值。根据tm值判断抑制剂与n39所形成螺旋结构的稳定性。
cd测试结果见图6所示,可见t-20可以与n39相互作用,所形成的复合物螺旋含量为48.6%,tm值为43.9℃;但是t20-trm与n39的相互作用极弱,导致仪器无法测定tm值,再次证明trm对t-20的重要性。脂肽lp-40与n39的相互作用明显增强,其复合物的螺旋含量为57.7%,tm值为51.3℃。连接臂的加入导致其螺旋含量有所减少,但多数对tm值影响不大,唯一tm值明显下降为含有最长连接臂(peg12)的lp-45。令人惊奇的是,导入ee**kk离子对极大地增强了脂肽的结合稳定性,表现为lp-50/n39复合物的tm值升高到63.3℃;而hiv-2/siv氨基酸的加入能够进一步增强脂肽的结合能力,使lp-51/n39和lp-52/n39复合物的tm值分别为72℃和79.1℃,它们比t-20和lp-40复合物的tm值显著增加,如图7所示。因此,三个强效抗病毒脂肽能够与靶序列形成极其稳定的螺旋结构,尤其是lp-52。相比较而言,lp-52/n39还具有最高的螺旋含量(63.8%)。
本实施例发现,脂肽的c端或n端截短均能不同程度地影响它们的结合能力,有的表现为tm值的下降,有的表现为螺旋和tm值双双下降。c端截短的脂肽(从lp-53到lp-59)的tm值显著下降,说明c末端的三个氨基酸(lek)在脂肽与nhr结合过程中的重要性。值得注意的是,具有强效抗病毒活性的lp-55和lp-56的tm值也有明显下降(从79.1到63.1℃),但比lp-53、lp-54、lp-57、lp-58和lp-59要高得多。特别是lp-58和lp-59由于较低的螺旋含量已不能确定tm值。n端截短脂肽(从lp-60到lp-68)的tm值也显著下降。基于lp-52的n端截短确实影响脂肽(lp-62到lp-65)的结合稳定性,但其复合物的tm值仍大于70℃,说明其仍具有极强的结合能力,这可能是它们保有强效的抗病毒能力的原因。值得注意的是,仅含24个氨基酸序列的脂肽lp-65亦具有较高的螺旋(63%)和tm值(72.1℃),而进一步的截短则严重影响其结合能力,如lp-66、lp-67和lp-68,这与它们的抗病毒活性相一致。相比较而言,去除c端3个氨基酸(lek)对结合稳定性(tm值)的影响要比去除n端1-4个氨基酸(weqk)的影响明显,说明脂肽c端在与作用靶点结合中起到更为重要的作用;而c端和n端均去除即核心序列脂肽lp-69结合稳定性显著下降,其tm值为51℃,比lp-52要低28.1℃。
另一比较有意思的现象是,n末端延长的脂肽其结合tm值有所提高,如lp-70到lp-75的表现,这与其抗病毒活性下降不一致。其中需要指出lp-74和lp-75包含了nhr口袋结合区(pbd)和m-t钩子基序,这使其不能与n39完全匹配。
该实施例结果还表明,硬脂酸(c18)修饰的脂肽lp-80同样具有极强的n39结合稳定性(tm值=79℃),但链长较短的脂肪酸c12修饰的脂肽lp-81和c8修饰的脂肽lp-82的螺旋含量和结合能力均明显下降,tm值分别为74.1℃和65.1℃。相对而言,它们的抗病毒活性下降更为明显(见图2)。胆固醇修饰的脂肽lp-83和lp-86、维生素e修饰的脂肽lp-85、二氢鞘氨醇修饰的脂肽lp-84和lp-87均具有很强的螺旋结合稳定性,这同它们的抗病毒活性相一致。同样,基于lp-80的n端截短脂肽(lp-88、lp-89、lp-90)的结合能力也比较强,tm值分别为76.5℃、70℃和71.1℃;但其c端lek去除的脂肽lp-91以及两端均截断的核心序列脂肽lp-92的螺旋稳定性则下降明显,其tm值分别为61℃和55.1℃。
该实施例通过大量的实验结果揭示了抑制剂的序列结构、结合稳定性和抗病毒活性彼此之间的相互关系,对了解本发明强效脂肽的作用机制提供了重要信息。虽然抑制剂的结合能力有时与其抗病毒活性不够一致,但总的来说本发明所述强效脂肽均具有极高的tm值。该实施例也揭示脂肽的抗病毒活性即取决于多肽序列,也取决于亲脂性化合物的性质。
实施例8.强效hiv膜融合抑制剂自身二级结构分析
为进一步探讨强效抗hiv脂肽的作用机制,本发明采用圆二色谱技术分析了t-20和代表性脂肽在溶液中的二级结构特点,方法与上述实施例7相同。为便于分析,分别测定抑制剂在10μm、20μm和40μm浓度下的α-螺旋含量和tm值(pbs溶液)。结果如图8所示,t-20在三种浓度条件下均表现为不规则的无序结构,lp-40在20μm和40μm时呈现微量螺旋结构,而四个强效脂肽(lp-50、lp-51、lp、52、lp-80)则表现为明显的螺旋结构,其中以lp-80的螺旋含量和tm值最高。因此,本发明的强效脂肽自身即可形成典型的螺旋结构,这与t-20显著不同。
实施例9.强效脂肽lp-80在大鼠的药代动力学分析
通过综合上述大量的研究结果可见,lp-80是本发明强效脂肽中体内外抗病毒活性较高并且非常稳定的脂肽。本实施例以lp-80为代表分析其在sd大鼠体内的药代动力学特征。试验共用12只sd大鼠,年龄为5-8周,体重182-219克。分为静脉注射组和皮下注射给药组,每组6只动物,雌雄各半。lp-80的给药剂量均为6毫克/公斤体重(mg/kg),lp-80用无菌蒸馏水溶解。各组动物于给药前、给药结束后5分钟、15分钟、30分钟、1小时、2小时、4小时、8小时、24小时、48小时、72小时、96小时、120小时、168小时和216小时采集血清样本。采用液相色谱质谱联用(lc-ms/ms)方法定量测定大鼠血清中lp-80的浓度,定量下限为1纳克/毫升(ng/ml)。采用非房室模型(nca)计算代谢动力学参数。试验结果见图9。lp-80静脉注射组和皮下注射给药组的平均末端消除半衰期(t1/2)分别为6.04小时和6.28小时。但值得特别指出的是,lp-80在静脉注射和皮下注射3天(72小时)后在血清中的浓度仍分别为7.75ng/ml和6.86ng/ml,换算成摩尔浓度则分别是2021.12pm和1789.02pm,该浓度分别是lp-80抑制hiv-1毒株nl4-3和jrcsf活性ic50值(2pm)的1010.56倍和894.51倍。该结果从药代动力学角度证实了上述实施例6中lp-80在猴体内的强效和长效的抗病毒能力。
实施例10.强效脂肽lp-80在猴子艾滋病模型的治疗效果评价
本实施例进一步考察了lp-80对猴子艾滋病感染模型的治疗效果,技术路线参考本发明人已公开发表的评价lp-19时所采用的方法(即背景技术参考文献30)。实验采用6只成年中国恒河猴(编号为a到f),雌雄各半,通过筛选确定siv、b型疱疹病毒(herpesvirusb)、猴淋巴细胞白血病病毒(simiant-lymphotropicvirus)为阴性;shiv毒株sf162p3由美国nih艾滋病试剂和参照物项目提供,在猴子外周血淋巴细胞(pbmc)扩增并测定tcid50剂量。通过静脉途径接种1000tcid50剂量sf162p3病毒到猴子体内建立感染,并定时测定猴子血浆病毒载量(rna拷贝数/毫升)变化情况。在猴子感染sf162p3后第197天通过皮下途径注射lp-80(用无菌蒸馏水溶解)进行治疗,lp-80按每公斤体重2毫克(2mg/kg)给药。每天一次给药治疗2周,然后改为四天一次治疗4周。在设定的时间点采取猴子血液分离血浆标本,采用实时定量逆转录聚合酶链反应技术(qrt-pcr)测定血浆中病毒载量(rna拷贝数/毫升)。按常规方法提取血浆rna并通过逆转录反应合成cdna标本;pcr引物针对siv的gag477(上游引物为gcagaggaggaaattacccagtac;下游引物为caattttacccaggcatttaatgtt;检测探针为fam-acctgccattaagcccga-mgb)。所用pcr仪器为peabi7500。检测方法灵敏度为每毫升血浆标本100rna拷贝。
实验结果见图10所示,开始治疗后第4天6只猴子中就有3只病毒载量已下降到检测线水平以下;治疗第8天后已有5只猴子测不到病毒载量;到第14天所有6只猴子测不到病毒载量。随后的1次/4天给药治疗期间所有猴子病毒载量均控制在检测线水平以下。于停药后第4天未见病毒反弹;停药后第10天其中有1只猴子(猴子a)出现病毒反弹;停药后第17天除猴子c外其余5只猴子均出现病毒反弹;到停药后24天所有猴子病毒载量出现反弹。该结果说明lp-80强大的抗病毒治疗效果。
序列表
<110>中国医学科学院病原生物学研究所
<120>艾滋病病毒膜融合抑制剂脂肽及药物用途
<160>12
<170>patentinversion3.5
<210>1
<211>28
<212>prt
<213>人工序列(artificialsequence)
<400>1
trpgluglnlysileglugluleuleulyslysalaglugluglngln
151015
lyslysasngluglugluleulyslysleuglulys
2025
<210>2
<211>28
<212>prt
<213>人工序列(artificialsequence)
<400>2
leuglualaasnileglugluleuleulyslysalaglugluglngln
151015
lyslysasngluglugluleulyslysleuglulys
2025
<210>3
<211>28
<212>prt
<213>人工序列(artificialsequence)
<400>3
tyrthrserleuileglugluleuilelyslysserglugluglngln
151015
lyslysasngluglugluleulyslysleuglulys
2025
<210>4
<211>27
<212>prt
<213>人工序列(artificialsequence)
<400>4
gluglnlysileglugluleuleulyslysalaglugluglnglnlys
151015
lysasngluglugluleulyslysleuglulys
2025
<210>5
<211>26
<212>prt
<213>人工序列(artificialsequence)
<400>5
glnlysileglugluleuleulyslysalaglugluglnglnlyslys
151015
asngluglugluleulyslysleuglulys
2025
<210>6
<211>26
<212>prt
<213>人工序列(artificialsequence)
<400>6
serleuileglugluleuilelyslysserglugluglnglnlyslys
151015
asngluglugluleulyslysleuglulys
2025
<210>7
<211>25
<212>prt
<213>人工序列(artificialsequence)
<400>7
lysileglugluleuleulyslysalaglugluglnglnlyslysasn
151015
gluglugluleulyslysleuglulys
2025
<210>8
<211>24
<212>prt
<213>人工序列(artificialsequence)
<400>8
ileglugluleuleulyslysalaglugluglnglnlyslysasnglu
151015
glugluleulyslysleuglulys
20
<210>9
<211>24
<212>prt
<213>人工序列(artificialsequence)
<400>9
ileglugluleuilelyslysserglugluglnglnlyslysasnglu
151015
glugluleulyslysleuglulys
20
<210>10
<211>25
<212>prt
<213>人工序列(artificialsequence)
<400>10
trpgluglnlysileglugluleuleulyslysalaglugluglngln
151015
lyslysasngluglugluleulyslys
2025
<210>11
<211>29
<212>prt
<213>人工序列(artificialsequence)
<400>11
trpgluglnlysileglugluleuleulyslysalaglugluglngln
151015
lyslysasngluglugluleulyslysleuglulyscys
2025
<210>12
<211>29
<212>prt
<213>人工序列(artificialsequence)
<400>12
leuglualaasnileglugluleuleulyslysalaglugluglngln
151015
lyslysasngluglugluleulyslysleuglulyscys
2025
- 还没有人留言评论。精彩留言会获得点赞!