一种基于二代测序技术检测同源重组通路基因突变的方法及试剂盒与流程
本发明涉及一种基于二代测序技术检测同源重组通路基因突变的方法及试剂盒,属于分子检测
技术领域:
。
背景技术:
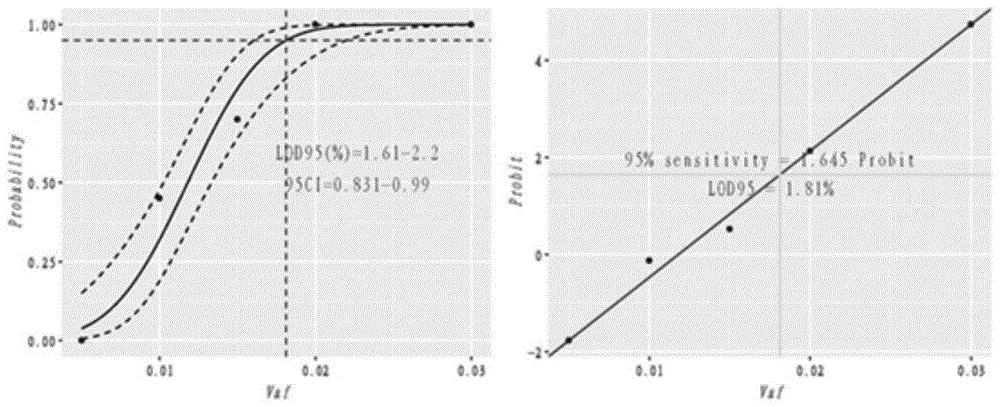
:同源重组(homologousrecombination)是指发生在非姐妹染色单体之间或同一染色体上含有同源序列的dna分子之间或分子之内的重新组合。同源重组可以使受损的染色体通过与另一条未受损染色体的相同dna进行自身的修复,这种修复保证了基因组的完整性。当细胞出现同源重组基因突变,导致同源重组缺陷(homologousrecombinationdeficiency,hrd)时,细胞不能通过同源重组的方式对dna进行自身修复。比如我们熟知的乳腺癌肿瘤相关基因brca1和brca2就是同源重组蛋白。当个体的brca1和brca2两基因发生突变时,其乳腺癌终生风险为87%,患卵巢癌的风险也超过40%,而且会导致发病年龄提前。聚腺苷二磷酸核糖聚合酶(polyadp-ribosepolymerase,parp)是一种dna修复酶,在dna修复通路中起关键作用。dna损伤断裂时会激活parp,它作为dna损伤的一种分子感受器,具有识别、结合到dna断裂位置的功能,进而激活、催化受体蛋白的聚adp核糖基化作用,参与dna的修复过程。因此,parp抑制剂是一种靶向聚adp核糖聚合酶的癌症疗法,它能将导致同源重组修复功能缺陷的肿瘤细胞发生合成致死,但不影响正常细胞,由此产生高选择性抗肿瘤作用。研究发现携带brca突变的肿瘤细胞对parp抑制剂的敏感度高。同源重组修复通路非常复杂,其突变的检测存在一定难度,因此建立准确、可靠、灵敏的检测方法具有十分重要的意义。但目前的同源重组通路基因检测方法中未能实现基因的全部覆盖,当前的检测方法未考虑同源重组基因的内含子区域,更没有考虑同源重组通路上的其他基因。如在检测brca1、brca2基因突变的方法中,同源重组修复基因的靶向测序主要涉及brca1、brca2基因的外显子区域,并没有考虑到brca1、brca2的内含子区域以及通路上的其他基因。brca1、brca2的外显子长度40nt到4931nt之间,内含子长度在92nt到14544nt之间。brca1、brca2上长度小于50nt的大片段缺失已经证明可以影响brca1、brca2的正常功能。对于长度较短且两侧内含子过长的外显子,捕获整个基因优于仅捕获外显子区域,增加了检测的准确性。另外研究表明同源重组通路上的atm基因具有细胞周期阻滞以及dna损伤后修复的功能,atm蛋白已经成为parp抑制剂的靶点之一。rad51产生的蛋白与单链和双链dna结合,催化同源dna间的识别和链交换,在同源重组修复过程中发挥重要功能。rad51foci无法形成是同源重组修复通路失效细胞的特征之一。每一个同源重组修复基因失效都有可能导致基因组不稳定的发生。基于此,本发明提供一种基于二代测序技术检测同源重组通路基因突变的方法及试剂盒。技术实现要素:现有技术在检测同源重组修复通路基因的突变时存在如下不足:1.现有技术在检测同源重组缺陷时,并没有考虑同源重组通路上的其他基因。2.现有产品没有加入突变特征作为辅助判断同源重组修复通路功能活性的方法。3.现有产品没有加入双等位基因致病突变负荷的计算,因此造成灵敏度较低,特异性较差。本发明提供一种基于二代测序技术检测同源重组通路基因突变的方法及试剂盒。本发明提供一种同源重组修复基因致病点突变及插入缺失突变、同源重组修复基因双等位基因致病突变负荷、同源重组修复基因突变特征、同源重组修复基因拷贝数变异、同源重组修复基因拷贝数负荷、中的一个或者多个的综合值来判断患者是否存在同源重组功能缺失的检测试剂盒。所述同源重组修复基因致病点突变及插入缺失突变、同源重组修复基因双等位基因致病突变负荷、同源重组修复基因突变特征、同源重组修复基因拷贝数变异、同源重组修复基因拷贝数负荷通过利用相关的靶向基因设计的探针(gene-panel)捕获的肿瘤样本以及正常样本测序得到。所述对肿瘤样本同源重组修复基因突变特征的计算主要通过同源重组基因靶向测序数据统计,主要步骤包括:1)对snv进行识别并注释到基因水平;2)根据注释结果进行筛选;3)使用软件(sigma)进行突变特征分析。其中,同源重组修复基因致病点突变及插入缺失突变的识别通过同源重组靶向基因测序统计得到。所述同源重组修复基因双等位基因致病突变负荷的计算主要通过统计同源重组修复基因的拷贝数以及等位基因频率得到。所述同源重组修复基因拷贝数负荷为通过计算每个超出阈值拷贝数变化的基因的数量。一种基于二代测序技术检测同源重组通路基因突变的方法,其包括如下步骤:a)将ffpedna和bcdna浓度稀释至6ng/μl,使用covarism220打断仪对片段进行处理得到片段化dna;b)将片段化dna加入到末端修复加a反应体系进行第一轮pcr反应,得到末端加a的片段化dna;向反应完毕的pcr反应体系中加入配置好的连接体系进行第二轮pcr反应,反应完毕后使用磁珠纯化得到纯化后的连接产物;c)向纯化后的连接产物加入配置好的扩增体系,按照98℃45sec,1循环;98℃15sec,60℃30sec,72℃30sec,n循环,72℃1min,1循环的反应程序进行pcr反应,8℃下终止反应,使用磁珠纯化后得到dna文库;使用qubit定量使得ffpedna文库≥350ng,bcdna文库≥200ng,并用2100生物分析仪对文库分析,主峰应位于150~500bp之间;d)杂交捕获:将ffpedna文库和bcdna文库按照质量比1:1进行混合,向混合后的dna文库加入人类cotdna和封阻序列,使用真空离心浓缩仪将dna文库蒸干;e)向蒸干后的dna文库加入dna杂交体系,震荡混匀离心后室温孵育,按照95℃30s,65℃4h的杂交反应条件进行杂交,即可得到捕获后的文库;f)捕获后文库扩增:向捕获后的文库中加入文库扩增反应体系,按照98℃45sec,1循环;98℃15sec,60℃30sec,72℃30sec,14循环,72℃1min,1循环的反应程序进行pcr反应得到扩增产物;使用磁珠纯化扩增产物;g)文库测序:按照ffpedna文库和bcdna文库以6:1比例进行混合,测试试剂对应位置,使用nextseqcn500基因测序仪上机测序;使用软件对数据进行处理,去掉接头、引物以及低质量的序列;使用二代测序数据比对软件bwa将预处理所得gene-panel的原始数据比对到人类参考基因组上,得到每条序列的位置信息以及比对质量信息,然后使用软件(如picard)对比对得到的结果进行质量评估。上述所述的检测方法中,末端修复加a反应步骤中第一轮pcr反应中,反应体系中包括末端修复酶2μl,末端修复缓冲液10μl,无核酸酶水48μl,反应条件为20℃,30min;65℃,30min;4℃终止反应;第二轮pcr反应的反应体系包含dna连接缓冲液30μl,dna连接酶3μl,连接增强子0.5μl,接头2.5μl,无核酸酶水14μl;反应条件为20℃,15min,4℃终止反应。上述所述的检测方法中,所述步骤c)中纯化后的连接产物为ffpedna时,n为8;纯化后的连接产物为bcdna时,n为6;步骤c)中所述的扩增体系中包括纯化后的连接产物20μl,高保真热启动酶混合液25μl,文库扩增引物5μl。上述所述的检测方法中,所述的dna杂交体系包括杂交缓冲液2.7μl,杂交缓冲液增强子8.5μl,dna捕获探针4.5μl,无核酸酶水1.3μl。上述所述的检测方法中,所述的文库扩增反应体系中包括高保真热启动酶混合液25μl、文库扩增引物5μl和上一步洗脱的dna20μl。上述所述的检测方法中,待测序样本在测序前进行相应处理,处理方法包括如下步骤:混合后的待测序样本进行qubit定量和2100分析片段大小并计算出摩尔浓度,加入5μl4nm待测序样本和5μl0.2nnaoh融合,涡旋振荡混匀后短暂离心,室温变性5分钟,向其中加入990μlht1缓冲液终止变性,涡旋振荡混匀,短暂离心,将已变性的待测序样本稀释至合适的上机浓度后进行测序。上述所述的基于二代测序技术检测同源重组通路基因突变的方法,其还包括生信学分析,其具体包括如下步骤:a)同源重组基因snv以及indel的识别:使用mutect2软件对实施例1中得到的肿瘤样本以及正常样本二代测序gene-panel比对结果进行分析,识别肿瘤样本中的体细胞突变以及胚系突变,使用annovar软件对mutect2识别出来的体细胞突变以及胚系突变进行注释;b)同源重组基因拷贝数变异检测:使用cnvkit软件对实施例1中得到的肿瘤样本以及正常样本二代测序gene-panel比对结果进行分析,得到每个基因的拷贝数值,判定标准为:高于一定阈值的基因为扩增,低于一定阈值的基因为缺失;c.突变特征分数计算:对于实施例2中得到的snv的过滤结果使用sigma软件计算得到同源重组相关的突变特征打分;d)双等位基因致病突变负荷计算:对于实施例2中步骤2得到的snv以及indel的注释结果进行判断,对满足条件的同源重组修复基因进行计数得到双等位基因致病突变负荷e)同源重组基因拷贝数负荷计算:对于实施例中2中得到的拷贝数变化的基因的拷贝数进行判断,对于拷贝数大于基线样本平均拷贝数与三倍标准差之和或小于基线样本平均拷贝数与三倍标准差之差的基因进行计数,得到拷贝数负荷。其中步骤a)中满足以下标准的即为致病突变:1)覆盖到突变位点上的序列个数大于200条;2)突变的频率大于5%;3)clinvar中记为致病突变,或突变类型为fs、truncate、splice。其中步骤d)中双等位基因致病突变的具体判定标准如下:1)野生型的等位基因上发生了loh并伴随一个致病的胚系突变;2)同时发生致病的胚系突变以及致病的体细胞突变;3)野生型的等位基因上发生了loh并伴随一个致病的体细胞突变;4)两个不同的致病的体细胞突变。本发明还提供一种基于二代测序技术检测同源重组通路基因突变的试剂盒,所述每一单位的试剂盒中包括:1)末端修复&加a反应体系:末端修复缓冲液900μl;末端修复酶180μl;2)连接反应体系:dna连接缓冲液1350μl;dna连接酶270μl;连接增强子45μl;接头01~406μl;3)pcr扩增反应体系:文库扩增引物485μl;高保真热启动酶混合液1215μl;4)杂交捕获反应体系:dna捕获探针20μl;封阻序列14μl;人类cotdna35μl;杂交缓冲液120μl;杂交缓冲液增强子38μl;2×磁珠清洗缓冲液1050μl;10×清洗缓冲液1175μl;10×清洗缓冲液2105μl;10×清洗缓冲液3105μl;10×清洗缓冲液4210μl;链霉亲和素磁珠350μl;5)纯化反应:纯化磁珠15ml。本发明与现有技术相比,具有如下技术优势:1.本发明加入了同源重组修复通路上的部分其他基因的外显子区域,扩大了检测灵敏度以及检测范围。2.针对捕获测序的探针设计存在捕获效率低、均一性较差的问题,本发明选择的基因组区域更适合捕获测序,通过检测,能够获得更高的捕获效率以及更好的均一性。3.本发明加入突变特征作为辅助判断同源重组修复通路功能活性的检测方法。4.本发明在同源重组修复通路活性预测中创造性地加入了双等位基因致病突变负荷,增加了算法灵敏度和特异性。附图说明图1ffpe_dna_pool(snv)的probit回归曲线。图2ffpe_dna_pool(<10ntindel)的probit回归曲线。图3psc_dna_pool(snv)的probit回归曲线。图4her通路假基因位点变异的检测相关相关性。具体实施方式以下通过具体实施例进一步描述本发明,但本领域技术人员应能理解,所述实施例并不以任何方式限定本发明专利保护的范围。实施例1.本申请试剂盒的肿瘤样本和血细胞样本基因序列的获得1.ffpegdna和bcgdna片段化处理ffpedna和bcdna浓度稀释至6ng/μl。取55μl进行打断,打断仪推荐使用covarism220,按下表设置设备参数进行片段化参数体积:55μl/样品targetbp(peak)200peakincidentpower(w)75dutyfactor10%cyclesperburst200treatmenttime(s)270s(ffpedna)/6min(bcdna)2.文库构建2.1末端修复&加a:2.1.1按下表配置末端修复&加a反应体系,振荡混匀,短暂离心。组分体积(μl)片段化dnax末端修复酶2末端修复缓冲液10无核酸酶水补足48总体积602.1.2将配置好的反应体系置于pcr仪上,按下表进行pcr反应。注意:pcr仪热盖温度设为85℃。2.2接头连接:2.2.1按下面表格配制连接体系,加入到上述产物中。振荡混匀,短暂离心。2.2.2将配置好的连接反应体系置于pcr仪上,按下表进行pcr反应。注意:pcr仪热盖温度设为关闭且连接反应完成后,应立即进行下一步。步骤温度时间接头连接20℃15min终止4℃∞2.3磁珠纯化步骤:2.3.1纯化磁珠使用前置于多用途旋转摇床,室温平衡至少30min。2.3.2在上述孵育结束的每个样本中加入55μl纯化磁珠,移液器吹打混匀。2.3.3室温孵育5min,然后转移至磁力架上约5min,直至溶液变澄清,小心弃除上清。2.3.4将离心管保持在磁力架上,加入200μl80%乙醇,静置30s,弃除全部上清。2.3.5重复上述步骤,用10μl枪头将残留液体彻底吸干。2.3.6干燥磁珠2-3min,待酒精挥发完全(正面看不在反光,背面看已经干燥),加入21μllowte缓冲液,吹打混匀。2.3.7室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取20μl上清至另一个新的8联排管中。2.4文库扩增:2.4.1按下面表格配制扩增体系,加入到连接产物中。振荡混匀,短暂离心。组分体积(μl)纯化后的连接产物20高保真热启动酶混合液25文库扩增引物5合计502.4.2短暂离心,置于pcr仪上,按下表进行pcr反应扩增反应条件:样本类型与扩增循环数:样本类型循环数ffpedna8bcdna62.5文库纯化:2.5.1纯化磁珠使用前置于多用途旋转摇床,室温平衡至少30min。2.5.2在上述扩增后的样本中加入25μl纯化磁珠,移液器吹打混匀。2.5.3室温孵育5min,然后转移至磁力架上约5min,直至溶液变澄清,吸取上清。2.5.4将上清转移至25μl纯化磁珠,移液器吹打混匀。2.5.5室温孵育5min,然后转移至磁力架上约5min,直至溶液变澄清,弃上清。2.5.6将离心管保持在磁力架上,加入200μl80%乙醇,静置30s,弃除全部上清。2.5.7重复上述步骤,用10μl枪头将残留液体彻底吸干。2.5.8干燥磁珠2-3min,待酒精挥发完全(正面看不在反光,背面看已经干燥),加入51μllowte缓冲液,吹打混匀。2.5.9室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取50μl上清至1.5ml离心管中。2.6文库质控:qubit定量:ffpedna文库≥350ng,bcdna文库≥200ng,用2100生物分析仪分析文库大小,主峰应位于150~500bp之间。3.杂交捕获3.1实验前准备:3.1.1打开真空离心浓缩仪,待温度升至60℃备用。3.2文库混合:3.2.1每种样品类型文库分别按下表格捕获质量计算体积加入1.5ml低吸附离心管中。文库类型捕获量(ng)/文库捕获样本数量总量(μg)ffpedna83121bcdna831213.2.2往上述混合好的dna文库中加入下述两个试剂,混匀,瞬时离心。3.2.3用封口膜封住ep管,用小枪头在膜上扎若干小孔。放入真空离心浓缩仪中蒸干(60℃,约20min-1h)。注:随时查看是否已蒸干。3.3变性与杂交:3.3.1配制dna杂交体系,如下表格所示。组分体积(μl)杂交缓冲液2.7杂交缓冲液增强子8.5dna捕获探针4.5无核酸酶水1.33.3.2充分混匀,瞬时离心,向蒸干后的dna文库管里中分别加入17μl上述配制好的杂交体系。3.3.3充分震荡混匀,短暂离心,室温孵育5min。3.3.4重复上述步骤。3.3.5将上述步骤中的液体转移至200μlpcr管中,置于pcr仪上,65℃杂交,杂交时间16h,杂交程序如下。3.4杂交后纯化:3.4.1配制清洗工作液:每个捕获体系所需缓冲液的配制方法如下表。组分组分/μl水/μl1×工作液体积/μl2×磁珠清洗缓冲液15015030010×清洗缓冲液12522525010×清洗缓冲液21513515010×清洗缓冲液31513515010×清洗缓冲液4302703003.4.2每个capture分装300μl清洗缓冲液4及100μl清洗缓冲液1至八连排中。3.4.3清洗缓冲液4和清洗缓冲液1使用前应于65℃孵育(热盖温度70℃)约45min。3.4.4链霉亲和素磁珠使用前,将其室温平衡30min。3.4.5取50μl链霉亲和素磁珠于八连排中,加入100μl磁珠清洗缓冲液,震荡混匀。置于磁力架上1min至液体澄清,弃去上清。3.4.6重复2次,加入100μl磁珠清洗缓冲液,震荡混匀。置于磁力架上1min至液体澄清,弃去上清。3.4.7从磁力架取下八连排,短暂离心,再次置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体。3.4.8按下表格配制磁珠悬浮液,加入上述清洗好的磁珠里。组分体积(μl)杂交缓冲液2.7杂交缓冲液增强子8.5无核酸酶水5.83.4.9充分震荡混匀,短暂离心,转移至pcr管中,放置65℃(热盖温度70℃)pcr仪中孵育15min。3.4.10用枪测量捕获过夜的杂交液,保证捕获过夜的杂交液体积为17μl,防止损失。3.4.11将65℃孵育后的磁珠悬浮液转移至捕获过夜的杂交液中,移液器吹打混匀(整个孵育过程pcr管不得脱离65℃,所有混匀步骤均用移液器在65℃pcr仪上吹打混匀)。置于pcr仪中65℃孵育45min(pcr热盖温度设为70℃),每隔一段时间用枪吹打一次保证磁珠悬浮。时间间隔是11min、11min、11min和12min。3.4.12孵育完成后,将pcr管中的液体转移至八连排中,加入100μl65℃预热的清洗缓冲液1,移液器吹打混匀。置于磁力架上1min至液体澄清,弃去上清。3.4.13从磁力架取下八连排,快速短暂离心(防止温度降低太多),置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体。3.4.14加入150μl65℃预热的清洗缓冲液4,移液器吹打混匀,65℃孵育5min,置于磁力架上1min至液体澄清,弃去上清。3.4.15再次加入150μl65℃预热的清洗缓冲液4,移液器吹打混匀,65℃孵育5min,置于磁力架上1min至液体澄清,弃去上清。3.4.16从磁力架取下八连排,短暂离心,置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体。注:整个热洗过程,温度尽量不要低于65℃,磁力架可置于加热仪上。3.4.17加入150μl室温放置的清洗缓冲液1,振荡30s,静止30s,再振荡30s,静止30s(共2min),短暂离心,置于磁力架上1min至液体澄清,弃去上清。从磁力架取下八连排,短暂离心,置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体。3.4.18加入150μl室温放置的清洗缓冲液2,振荡30s,静止30s,再振荡30s,静止30s(共2min),短暂离心,置于磁力架上1min至液体澄清,弃去上清。从磁力架取下八连排,短暂离心,置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体。3.4.19加入150μl室温放置的清洗缓冲液3,振荡30s,静止30s,再振荡30s,静止30s(共2min),短暂离心,置于磁力架上1min至液体澄清,弃去上清。从磁力架取下八连排,短暂离心,置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体。3.4.20向离心管中加入21μl无核酸酶水洗脱,震荡混匀,进行下一步扩增试验。3.5捕获后文库扩增:3.5.1根据捕获个数,按照下表配制文库扩增反应体系,震荡混匀。组分体积(μl)高保真热启动酶混合液25文库扩增引物5上一步洗脱的dna203.5.2短暂离心,将文库扩增反应体系分装至pcr管中,每管30μl。置于pcr仪上,按下表进行pcr反应3.6扩增后纯化:3.6.1取出纯化磁珠,室温平衡30min备用。3.6.2取75μl纯化磁珠于1.5ml离心管中,加入50μl扩增后的捕获dna文库上清,振荡混匀,室温孵育10min。3.6.3置于磁力架上1min至液体澄清,弃去上清。3.6.4从磁力架取下1.5ml离心管,短暂离心,置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体。3.6.5加入200μl80%乙醇孵育30sec后弃去。注意:80%乙醇现用现配。重复用200μl80%乙醇清洗一次。3.6.6从磁力架取下1.5ml离心管,短暂离心,置于磁力架上,用10μl枪头彻底弃去离心管底部的残留液体,室温干燥至乙醇完全挥发(前面看磁珠不反光,背面看干燥)。注意:磁珠过分干燥dna产量会减少。3.6.7从磁力架取下离心管,加入40μl超纯水,振荡混匀。室温孵育2min。3.6.8短暂离心,置于磁力架上1min至液体澄清,将capture样本转入新的离心管中。3.7capture质检:分别取1μlcapture用于qubit浓度检测。4.文库测序4.1根据上机需求对捕获后文库进行混合,按照每个捕获后文库包含的子文库数进行混合,ffpedna文库和bcdna文库以6:1比例进行混合。capture类型文库个数加入量(ng)/文库总量(ng)ffpedna1212144bcdna142284.2对混合后的待测序样本进行qubit定量和2100分析片段大小,并计算出摩尔浓度。4.3将待测序样本加水稀释至4nm,体积约20μl。4.4将naoh溶液加水稀释至0.2n,体积约20μl。4.5取一个新的离心管,加入5μl4nm待测序样本和5μl0.2nnaoh融合,涡旋振荡混匀后短暂离心,室温变性5分钟。4.6加入990μlht1缓冲液终止变性,涡旋振荡混匀,短暂离心,此时待测序文库浓度为20pm。4.7按照下表将已变性的待测序样本稀释至合适的上机浓度:上机浓度稀释方法1.8pm117μl文库(20pm)+1183μlht14.8将上述文库加入测试试剂对应位置,用illumina公司生产的基因测序仪nextseqcn500上机测序。5.测序数据处理对于测序的下机数据,使用软件(如trimmomatic)对数据进行处理,去掉接头、引物以及低质量的序列。使用二代测序数据比对软件bwa将预处理所得以gene-panel的原始数据比对到人类参考基因组上,得到每条序列的位置信息以及比对质量信息。然后使用软件(如picard)对比对得到的结果进行质量评估。实施例2.本申请试剂盒同源重组基因致病的检测1.同源重组基因snv以及indel的识别使用mutect2软件对实施例1中得到的肿瘤样本以及正常样本二代测序gene-panel比对结果进行分析,识别肿瘤样本中的体细胞突变以及胚系突变,使用annovar软件对mutect2识别出来的体细胞突变以及胚系突变进行注释,满足以下标准的即为致病突变:1)覆盖到突变位点上的序列个数大于200条;2)突变的频率大于5%;3)clinvar中记为致病突变,或突变类型为fs、truncate、splice。2.同源重组基因拷贝数变异检测使用cnvkit软件对实施例1中得到的肿瘤样本以及正常样本二代测序gene-panel比对结果进行分析,得到每个基因的拷贝数值,判定标准为:高于一定阈值的基因为扩增,低于一定阈值的基因为缺失。实施例3.本申请试剂盒突变特征分数计算体细胞突变由不同的外界或内部因素诱导产生,其中包括dna复制机制的错误、内在或者外在因素的诱导、dna修饰酶的修饰或者dna修复酶失效等。不同因素导致的体细胞突变会有不同的突变类型的组合,就是所谓的突变特征。已有报道,突变特征中的signature3与同源重组通路缺陷有非常强的相关性。对于实施例2中得到的snv的过滤结果使用sigma软件计算得到同源重组相关的突变特征打分。实施例4本申请试剂盒双等位基因致病突变负荷计算1.双等位基因致病突变是指在肿瘤样本中同源重组基因的两个等位基因都失活。具体判定标准如下:1)野生型的等位基因上发生了loh并伴随一个致病的胚系突变;2)同时发生致病的胚系突变以及致病的体细胞突变;3)野生型的等位基因上发生了loh并伴随一个致病的体细胞突变;4)两个不同的致病的体细胞突变。2.对于实施例2中步骤2得到的snv以及indel的注释结果按照步骤1中的规则进行判断,对满足条件的同源重组修复基因进行计数得到双等位基因致病突变负荷。实施例5本申请试剂盒同源重组基因拷贝数负荷计算1.对于实施例中2中得到的拷贝数变化的基因的拷贝数进行判断,对于拷贝数大于基线样本平均拷贝数与三倍标准差之和或小于基线样本平均拷贝数与三倍标准差之差的基因进行计数,得到拷贝数负荷。实施例6本申请试剂盒的hrr通路基因的snv/indel阳性阈值的确定采用23例已知的brca1/2野生型的组织样本和36例brca1/2野生型的血细胞样本进行文库构建、靶向捕获、上机测序和测序数据处理,实验过程和测序数据处理参考实施例1,生信分析参考实施例2。以brca1/2基因上clinvar数据库中注释为pathogenic/likelypathogenic位点,包含573个snv位点与156个indel位点的检测特异性(阴性一致率)为标准,对突变检出的阳性阈值进行确认。2.实验步骤及测序数据处理参考实施例1,生信分析参考实施例2。3.实验结果实施例9本申请试剂盒的hrr通路基因最低检测限的检测1.制备hrr通路基因最低检测限标准品采用已知突变信息的ffpe样本的dna按照一定的比例混合,建立2个含有不同变异类型的ffpednapool,其中ffpe_dna_pool1中的变异类型为单核苷酸突变,ffpe_dna_pool2中含有的变异类型为<10nt的插入/缺失突变。同时将商业阳性标准品基因组psc与阴性标准品12878进行比例混合。hrr通路基因最检测限标准品突变信息如表1所示:表1:2.实验步骤及测序数据处理参考实施例1,生信分析参考实施例23.重复检测20次4.实验结果ffpe_dna_pool(snv)的probit回归曲线如图1所示.ffpe_dna_pool(<10ntindel)的probit回归曲线如图2所示。psc_dna_pool(snv)的probit回归曲线如图3所示。实施例7本申请试剂盒的hrr通路基因精密度的检测1.制备hrr通路基因精密度标准品采用不同突变类型的ffpe肿瘤组织样本,测试了批内重复性(intra-runrepeatability)以及批间重复性(inter-runreproducibility)。批内重复性是指在同一条件下同一批次内(试剂批号以及仪器完全一样),对样本进行平行操作时检测结果的可重复性。批间重复性是指在不同的操作中对同一样本的检测结果的可重复性,其中不同的条件包括了不同的操作人,不同的测序仪等,在本次研究中我们评估了不同批次操作以及不同的测序仪对批间重复性的影响。hrr通路基因精密度标准品信息如表2所示:表2:2.实验步骤及测序数据处理参考实施例1,生信分析参考实施例23.重复检测10次4.实验结果实施例8本申请试剂盒的hrr通路基因准确性的检测1.制备hrr通路基因准确性标准品采用不同突变类型的ffpe肿瘤组织样本,分析本产品与本公司ldt产品检测brca1/2以及hrr通路上的其他变异和基因snv/indel变异类型的一致性。hrr通路基因准确性标准品信息如表3所示:表3:2.实验步骤及测序数据处理参考实施例1,生信分析参考实施例23.实验结果her通路假基因位点变异的检测相关相关性如图4所示。经过panelwise位点分析两平台相关性很好,y=1.0338x-0.0043且r2=0.9963。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1