一种高纯度稳定晶型的醋酸艾司利卡西平的合成方法与流程
1.本发明属于医药化工领域,涉及高纯度醋酸艾司利卡西平的合成方法及其稳定晶型。
背景技术:
2.醋酸艾司利卡西平(eslicarbazepine acetate,1)化学名为(s)-10-乙酰氧基-10,11-二氢-5h-二苯并[b,f]氮杂-5-甲酰胺,是由sunovionpharma公司开发的s-利卡西平的醋酸酯前药,s-利卡西平是奥卡西平的主要活性代谢产物,能够阻断电压依赖性na
+
通道,与奥卡西平相比,醋酸艾司利卡西平的耐受性更好。醋酸艾司利卡西平于2009年在欧洲上市,商品名为zebinix,2013年11月美国fda批准该药用于成人癫痫部分性发作的治疗。目前,该药还未在中国上市,本文作者对一种稳定晶型的醋酸艾司利卡西平的合成工艺进行研究。
[0003]
文献(醋酸艾司利卡西平的合成工艺改进[j].中国药物化学杂志,2016,26(01):33-36.)报道了以奥卡西平为起始原料,与硼氢化钠反应得到利卡西平,经手性拆分水解、酰化成酯得到醋酸艾司利卡西平。此路线初始原料奥卡西平的市场价格昂贵,生产成本很高,且手性拆分操作大大降低了产物的收率,无实际应用价值。
[0004]
文献(醋酸艾司利卡西平的合成工艺改进[j].中国药物化学杂志,2016,26(01):33-36.)报道了以奥卡西平为起始原料,经ruc
l2
不对称还原、酰化成酯得到醋酸艾司利卡西平。此路线中还原反应的副产物很难除去,需经柱色谱分离纯化,无法实现工业化生产,而且所用手性催化剂ruc
l2
成本很高、用量较大,对终产物还会造成重金属残留的隐患。
技术实现要素:
[0005]
本发明的目的在于提供一种醋酸艾司利卡西平的稳定晶型及其合成方法,原料易得,合成步骤简单,成本低收率高,生产过程安全,对环境无污染。且生产出的产品为一种稳定晶型,并且具有较好的理化性质,纯度高,最大杂质小于0.05%,在高温,高湿,光照条件下均稳定。
[0006]
本发明解决其技术问题所采用的技术方案为:提供一种醋酸艾司利卡西平的稳定晶型及其合成方法。
[0007]
具体而言,第一方面,本发明提供了醋酸艾司利卡西平的一种稳定晶型,其特征在于,在使用cu-kα辐射,以2θ角度表示的x-射线衍射在5.060、5.600、11.180、16.819、19.340、21.860有衍射峰。
[0008]
在第一方面,本发明也提供了醋酸艾司利卡西平的一种稳定晶型,其特征在于,所述的醋酸艾司利卡西平晶型具有如图1所示的x-射线衍射图谱。
[0009]
进一步地,更优选本发明第一方面的醋酸艾司利卡西平晶型,具有如图2所示的热差分析(dsc)图谱,图2显示本发明第一方面的醋酸艾司利卡西平晶型,其差热分析曲线在188.2
±
2℃有一尖吸热峰。
[0010]
在第二方面,本发明提供了本发明第一方面醋酸艾司利卡西平晶型的合成方法,其特征在于,所述的合成方法按以下顺序进行:(1)将10-甲氧基亚氨基芪在酮类有机溶剂中溶解并在酸性条件下脱保护得到中间体i;(2)采用烷类、醚类有机溶剂将步骤(1)生成的溶液进行不对称还原得到中间体ii;(3)将步骤(2)所得溶液乙酰化成酯,再进行酰胺化得到醋酸艾司利卡西平。
[0011]
优选在本发明第二方面的合成方法中,步骤(1)中的酮类有机溶剂是丙酮、甲乙酮和/或戊酮,优选丙酮。
[0012]
也优选在本发明第二方面的合成方法中,步骤(1)脱保护反应中反应温度为20~60℃,优选30~40℃。
[0013]
更优选在本发明第二方面的合成方法中,步骤(1)脱保护反应中使用的酸为盐酸、硫酸和/或醋酸,优选5%~15%盐酸。
[0014]
优选在本发明第二方面的合成方法中,步骤(2)中烷类有机溶剂是二氯甲烷、氯仿、二氧六环、甲苯等,手性还原剂选用(r)-2-甲基-cbs-恶唑硼烷与硼烷二甲硫醚;或(r)-2-甲基-cbs-恶唑硼烷与硼烷四氢呋喃。
[0015]
也优选在本发明第二方面的合成方法中,步骤(2)不对成还原反应中底物与还原剂bh3。me2s的摩尔比为1:1.0~1:2.0,优选1:1.1~1:1.5,更优选1:1.3。
[0016]
也优选在本发明第二方面的合成方法中,步骤(2)不对成还原反应中底物与r-2-me-cbs摩尔比为1:0.1~1:1.0,优选1:0.1~1:0.5,更优选1:0.2。
[0017]
更优选在本发明第二方面的合成方法中,步骤(2)不对成还原反应中反应温度为-20~20℃,优选-10~5℃。
[0018]
优选在本发明第二方面的合成方法中,步骤(3)乙酰化反应中溶剂为二氯甲烷和/或乙酸乙酯,优选乙酸乙酯;有机碱为三乙胺,氨水和/或n、n-二异丙基乙基胺,优选三乙胺。
[0019]
或优选在本发明第二方面的合成方法中,步骤(3)乙酰化反应中中间体ii和三乙胺的摩尔比为1:1.0~5.0,优选1:2.0。
[0020]
也优选在本发明第二方面的合成方法中,步骤(3)乙酰化反应中中间体ii和乙酸乙酯的摩尔比为1:1.0~5.0,优选1:3.0。
[0021]
更优选在本发明第二方面的合成方法中,步骤(3)酰胺化反应中温度为-20~15℃,优选-5~5℃。
[0022]
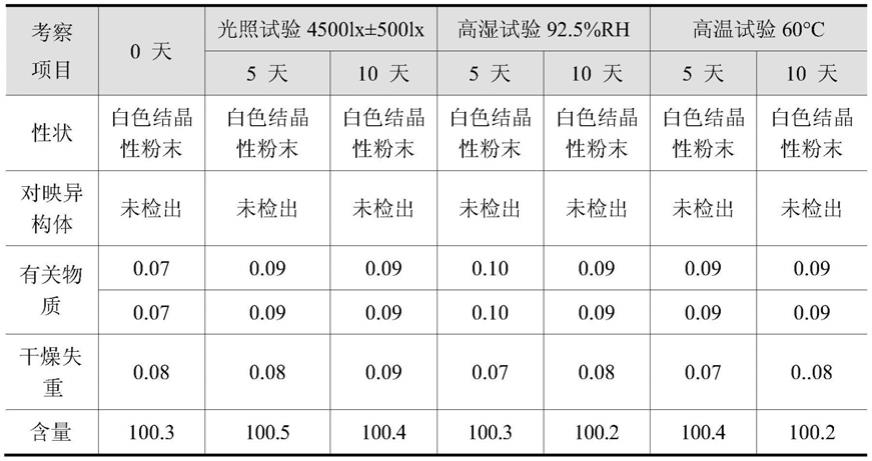
本发明合成工艺得到一种稳定晶型的醋酸艾司利卡西平,将任一晶型或无晶型醋酸艾司利卡西平在有机溶剂中甲醇、乙醇、异丙醇、乙酸乙酯中完全溶清,然后缓慢析出本发明第一方面的晶体。通过对晶型稳定性进行研究,考察条件为高温(60℃
±
2℃),高湿度(25℃,92.5%rh),强光照(4500
±
500lx),考察指标为性状、对映异构体、干燥失重、有关物质、含量。
[0023]
结果:样品经高温、高湿、光照破坏10天,其性状、对映异构体、有关物质、干燥失重及含量均无明显变化。说明化学稳定性良好,适合药物制剂的制造及长期储存。
[0024]
下面是合成醋酸艾司利卡西平的方程式:
[0025]
本发明与现有方法相比的有益效果是,获得了新的醋酸艾司利卡西平晶型,其被确认不含结晶水和结晶溶剂;其化学稳定性被证明良好;其原料易得,合成步骤简单,成本低收率高,生产过程安全,设备要求简单。
附图说明
[0026]
图1为本发明的醋酸艾司利卡西平晶型的x-射线衍射图谱。
[0027]
图2为本发明的醋酸艾司利卡西平晶型的dsc图谱。
具体实施方式
[0028]
下面结合实施事例对本发明进一步说明。
[0029]
实施例1:取10-甲氧基亚氨基芪43g,加入101g四氢呋喃,加入72g盐酸,加热至60~65℃,反应5h,加约258g自来水,搅拌10min,抽滤,得黄色沙状固体中间体i约38g,收率为95%。
[0030]
实施例2:取中间体i 25g,加入二氯甲烷,降温5~10℃,滴加bh2.me2s 95.g和r-2-me-cbs 33g,搅拌3h,加盐酸405g,分层,有机相减压浓缩得中间体ii 29g,收率75%。
[0031]
实施例3:取中间体i 25g,加入二氯甲烷,降温5~10℃,滴加bh2.me2s 85.g和r-2-me-cbs 28g,搅拌3h,加盐酸405g,分层,有机相减压浓缩得中间体ii 27g,收率70%。
[0032]
实施例4:取中间体ii 29g,加入乙酸乙酯191g,三乙胺28g,乙酸酐41g,升温回流,
反应2h,加水191g,分层。有机相降温至5℃,滴加clso2cno 19g,反应3h,滴加水380g,搅拌1小时,分液,有机相减压浓缩得类白色固体醋酸艾司利卡西平粗品24g,收率:57%。(es,m/z):[m+1]
+
=297,1h-nmr:(600mhz,cdcl3)δ:2.08(d,j=6.6hz,3h),3.10(dd,j=13.8,6.6hz,1h),3.59(d,j=13.8hz,1h),5.40(brs,2h),5.98~6.39(m,1h),7.18~7.30(m,6h),7.41~7.45(m,2h)。
[0033]
实施例5:取醋酸艾司利卡西平粗品25g,加入甲醇60g,升温回流,加入活性炭1.5g回流30min,热过滤,降温析晶,过滤得类白色粉末状固体醋酸艾司利卡西平15.6g,收率80%。
[0034]
实施例6:取醋酸艾司利卡西平粗品25g,加入乙酸乙酯60g,升温回流,加入活性炭1.5g回流30min,热过滤,降温析晶,过滤得类白色粉末状固体醋酸艾司利卡西平16g,收率82%。
[0035]
实施例7:取醋酸艾司利卡西平粗品25g,加入异丙醇60g,升温回流,加入活性炭1.5g回流30min,热过滤,降温析晶,过滤得类白色粉末状固体醋酸艾司利卡西平15.8g,收率81%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1