一种阿昔洛韦中间体N(2),9-二乙酰鸟嘌呤的制备方法与流程
本发明属于洛韦类抗病毒药中间体制备技术领域,具体是涉及一种阿昔洛韦中间体n(2),9-二乙酰鸟嘌呤的制备方法。
背景技术:
洛韦类药物,主要包括阿昔洛韦、更昔洛韦、伐昔洛韦与法昔洛韦等,为广谱的抗病毒药。在临床上用于治疗单纯疱疹及与hsv感染相关的疾病,如多形性红斑、带状疱疹、首发与复发性生殖器疱疹、hsv脑炎,以及水痘、带状疱疹和vzv脑炎、爱滋病、器官移植、恶性肿瘤病人严重的cmv感染所致的肺炎、肠炎及视网膜炎等。
n(2),9-二乙酰鸟嘌呤的合成方法,传统的合成方法主要是以鸟嘌呤或者鸟嘌呤核苷为原料进行乙酰化反应制备,该工艺也已经工业化生产也已经几十年了,工艺也比较稳定,公开报道的文献也比较多。文献(pulido,daniel;sanchez,albert;robles,jordi;pedroso,enrique;grandas,annaeuropeanjournaloforganicchemistry,2009,#9p.1398-1406)和文献(chemicalandpharmaceuticalbulletin,,vol.36,#3p.1153-1157)以及专利(us4423050a1,us4565868a1,us5250535a1和us4355032a1)均报道了以鸟嘌呤为起始原料,用乙酰化试剂(如醋酐或乙酰氯等)进行酰化反应制备n(2),9-二乙酰鸟嘌呤(ⅳ)。该方法工业化生产比较成熟,产品收率也很可观,但是产品质量较差,纯度很难超过98%,外观也较差。其合成路线如下:
虽然文献(organicpreparationsandproceduresinternational,,vol.44,#4p.387-391)报道了以鸟嘌嘌呤核苷为起始原料,于大大过量的醋酐中回流反应24~36小时,制备n(2),9-二乙酰鸟嘌呤(ⅳ),虽然该方法制备的双乙酰鸟嘌质量和外观较前面的方法都有很大改善,但是其醋酐单耗较高、反应时间长和副产四乙酰核糖提纯回收工艺复杂等缺点也限制了该工艺的工业化生产的推广应用,其合成路线如下:
以上几种制备方法均会涉及起始原料鸟嘌呤(或鸟嘌呤核苷)的使用,而传统的鸟嘌呤合成工艺却是一个高能耗,高排放,高危工艺多的生产工艺。
鸟嘌呤传统的合成工艺中,都是以氰乙酸酯与硝酸胍环合后进行亚硝化得到2,4-二氨基-5-亚硝基-6-羟基嘧啶(本文简称亚硝嘧啶),再将亚硝嘧啶催化还原制备三氨基嘧啶硫酸盐,再经环合、脱色、精制得到鸟嘌呤。其合成路线如下:
该方法中存在以下几个问题:1)催化加氢还原用到的铂、钯、钌、铑和镍等金属催化剂为还原剂,这类催化剂要么价格昂贵,要么回收成本高;2)加氢还原工艺属于高危工艺,安全成本较高;3)工艺中环合反应时间都要24小时以上,生产周期较长;4)工艺用到了大量氢氧化钠、硫酸和盐酸以及需要大量活性炭脱色精制才能得到合格的鸟嘌呤产品,这样会产生大量的高盐强酸废水和“危废”,环保成本也大大增加。
专利ep415028、de-ps3729471、de-ps3638635、de-ps3638635、us5688947、cn1296945、cn102399194都是采用贵金属催化氢化法制备三氨基嘧啶硫酸盐;陈文华等在《广东化工》vol.39(24),p59~60中采用雷尼镍催化氢化法制备三氨基嘧啶硫酸盐。
另外一方面,现有的n(2),9-二乙酰鸟嘌呤的合成方法中,均需要在大大过量的醋酐中进行反应。反应结束后,溶剂中由于参杂了副产物乙酸,导致容易无法直接回用。导致大量酸性废液的产生,后处理成本较高。
技术实现要素:
本发明提供了一种阿昔洛韦中间体n(2),9-二乙酰鸟嘌呤制备的全新方法。
针对传统工艺中存在的产品质量偏低,安全风险高和“三废”量大等问题,本发明提供了一种绿色环保、安全风险低和产品收率高、质量优等优势的全新制备方法。
一种阿昔洛韦中间体n(2),9-二乙酰鸟嘌呤的制备方法,包括:
(1)将2,4-二氨基-6-羟基-5-甲酰胺基嘧啶溶于醋酸中,滴加醋酐醋酸溶液,同时进行选择性酰化反应,得到2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶;
(2)2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶在催化剂a作用下,进行环合反应,得到2-乙酰氨基-6羟基嘌呤;
(3)2-乙酰氨基-6羟基嘌呤在催化剂c作用下,于醋酐和醋酸混合溶剂中进行酰化反应,得到n(2),9-二乙酰鸟嘌呤。
作为进一步优选,一种阿昔洛韦中间体n(2),9-二乙酰鸟嘌呤的制备方法,包括下列步骤:
(1)将2,4-二氨基-6-羟基-5-甲酰胺基嘧啶(i)溶于醋酸中,在一定温度下,滴加一定含量的醋酐醋酸溶液进行酰化反应,反应毕,再减压浓缩蒸出溶剂后,得到2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶(ii),蒸馏回收的醋酸可反复套用;
(2)向步骤(1)得到的化合物(ii)中依次加入催化剂a和溶剂b,升温至回流进行环合反应,边回流边脱去反应产生的水,反应毕,降温结晶,过滤得到淡黄色固体粉末2-乙酰氨基-6羟基嘌呤(iii),母液中的溶剂蒸馏回收后可循环套用;
(3)将步骤(2)中得到的化合物(iii)和催化剂c溶于一定含量的醋酐醋酸溶液中,升温至一定温度下进行催化酰化反应,反应毕,降温结晶,过滤,干燥得到类白色固体粉末n(2),9-二乙酰鸟嘌呤(iv),过滤母液可根据检测醋酐含量或补加新醋酐循环套用,生成的副产醋酸也可蒸馏回收后套用至步骤(1)反应或作为副产品销售。
本发明阿昔洛韦中间体n(2),9-二乙酰鸟嘌呤的制备方法的合成路线如下所示:
作为优选,步骤(1)中,所述醋酐醋酸溶液中醋酐的含量为:50%~100%,进一步优选为70%~90%。步骤(1)中,反应结束后,减压浓缩蒸出溶剂后得到所述的2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶,溶剂直接作为醋酐醋酸溶液回用。当需要补加酸酐时,可根据需要补加。
作为优选,步骤(1)中加入的醋酐和2,4-二氨基-6-羟基-5-甲酰胺基嘧啶的摩尔比为(1~5):1。进一步优选为:醋酐/化合物(i)=(1.2~2.5)/1.0。
本发明采用首先利用醋酸溶解所述原料2,4-二氨基-6-羟基-5-甲酰胺基嘧啶,然后采用将醋酐醋酸溶液加入到体系中,进一步降低了反应体系中醋酐的浓度,避免二取代的发生,进一步提高步骤(1)中乙酰化的选择性。同时本发明采用滴加的方式加入,进一步提高了选择性。
作为优选,步骤(1)中,滴加醋酐醋酸溶液时间为3~10h。进一步优选为5~7h。滴加的同时体系进行反应。
作为优选,步骤(1)中,滴加或反应温度为50~100℃,进一步优选为60~70℃。较低的反应温度,也对于酰化反应的选择性提供了进一步保证。
步骤(2)中,所述催化剂a选自三乙胺、吡啶、哌啶、乙醇胺、醋酸钠、对甲苯磺酸、硫酸氢钠、氯化锌、氯化锡、四丁基硫酸氢铵和四丁基溴化铵中的一种或多种,进一步优选为醋酸钠、硫酸氢钠和四丁基溴化铵。其中催化剂a加入量为2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶加入量的1%~15%;进一步优选为5%~10%。
步骤(2)中采用的溶剂选自乙酸丁酯、辛醇、甲苯、二甲苯(邻,对,间二甲苯)中的一种或多种。进一步优选为甲苯或二甲苯。作为优选,其中溶剂b用量相对于化合物(ii)的用量为:溶剂b/化合物(ii)=(1~10)/1,进一步优选为:溶剂b/化合物(ii)=(3~6)/1。
作为优选,环合反应时间为10~24h,进一步优选为12~16h。
作为优选,环合反应温度为90~145℃,进一步优选为110~135℃。
步骤(1)中,为了进一步促进反应,反应过程中利用分水器进行分水,反应结束后冷凝至室温结晶,得到所述2-乙酰氨基-6羟基嘌呤。
步骤(3)中,所述催化剂c选自三乙胺、吡啶、哌啶、乙醇胺、醋酸钠、对甲苯磺酸和硫酸氢钠中的一种或多种;进一步优选为三乙胺和醋酸钠。
作为优选,其中催化剂c相对2-乙酰氨基-6羟基嘌呤的配比为:1%~15%。进一步优选为5%~10%。
作为优选,步骤(3)中,所述醋酐和醋酸混合溶剂中醋酐含量为50%~100%,进一步优选为65%~85%。
作为优选,步骤(3)中,溶剂用量相对化合物(iii)用量的倍数为5~20倍,进一步优选为6~15倍。
作为优选,步骤(3)的反应时间为5~20h,,进一步优选为8~15h。反应温度为120~140℃,进一步优选为125~135℃。
作为优选的技术方案,步骤(1)中,所述醋酐醋酸溶液中醋酐的含量为:70%~90%,其中纯醋酐和2,4-二氨基-6-羟基-5-甲酰胺基嘧啶的摩尔比为(1.2~2.5):1;反应温度为60~70℃;步骤(2)催化剂a为醋酸钠、硫酸氢钠和四丁基溴化铵中的一种或多种,反应温度为110~135℃;步骤(3)中,所述催化剂c为三乙胺和醋酸钠中的一种或两种,醋酐和醋酸混合溶剂中醋酐含量为65%~85%;酰化反应温度为125~135℃。
本发明中一种阿昔洛韦中间体n(2),9-二乙酰鸟嘌呤的制备方法的工艺,克服了传统工艺中普遍存在的安全风险高、产品质量不稳定、“三废”排放量大等问题。
与传统工艺技术相比,本发明的有益效果体现在:
(1)原辅料单耗低,几乎所有溶剂都可以回收循环利用,原料成本低;
(2)产品总摩尔收率高(第一步95%;第二步97%;第三步98%),总摩尔收率可达90%以上;
(3)产品质量稳定,纯度都能到达98%以上,质量和外观优于市场同类产品;
(4)无高危工艺,安全风险低;
(5)“三废”排放少,几乎没有任何废水产生,只有溶剂回收时的少量蒸馏残渣生成,绿色清洁环保。
与传统工艺相比,本发明合成工艺无大量水引入和大量的无机盐产生,所用溶剂均可实现回收利用,工艺过程简便已操作,产品收率略高于现有技术且质量符合相应的质量标准。因此本发明的制备方法是一种高效、经济、绿色环保的制备方法。
附图说明
图1为2-乙酰氨基-6羟基嘌呤的ms谱图。
图2为n(2),9-二乙酰鸟嘌呤的ms谱图。
图3为n(2),9-二乙酰鸟嘌呤的氢谱。
具体实施方式
为了使本发明的技术方案和优点更加清楚,下面将本发明的优选实施例子作进一步的详细描述,本发明包括但不限于以下实施例、及其所展示的工艺参数。
实施例1:
将50g2,4-二氨基6-羟基-5-甲酰胺基嘧啶(mw169.14,0.30mol)溶于100g醋酸(mw60.05,1.66mol)中,缓慢升温至60~70℃,控制温度60~70℃,缓慢滴加86g的含量80%醋酐醋酸溶液(mw102.09,0.67mol),滴加时间控制在6h。
滴毕,开始减压蒸馏回收醋酸溶剂,浓缩干后,得到浓缩残留固体约60.9g即为2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶(ⅱ),粗摩尔收率约96.1%,纯度98.2%。回收醋酸可反复套用。
向化合物(ⅱ)中加入5g催化剂a(醋酸钠)和300g二甲苯,开搅拌和加热,升温至125~135℃,回流脱水反应14h,当分分水器中没有明显水滴产生时,反应结束。开始降温至室温,保温结晶2小时,过滤,干燥得到淡黄色固体粉末约53.9g,即为2-乙酰氨基-6羟基嘌呤(ⅲ),摩尔收率96.9%,纯度98.6%。过滤母液可蒸馏回收后套用,这样既可以避免废液的产生,其中的催化剂可以继续发挥作用,进一步降低了催化剂的消耗。
本实施例制备得到的2-乙酰氨基-6羟基嘌呤(ⅲ)其结构分析数据如下:
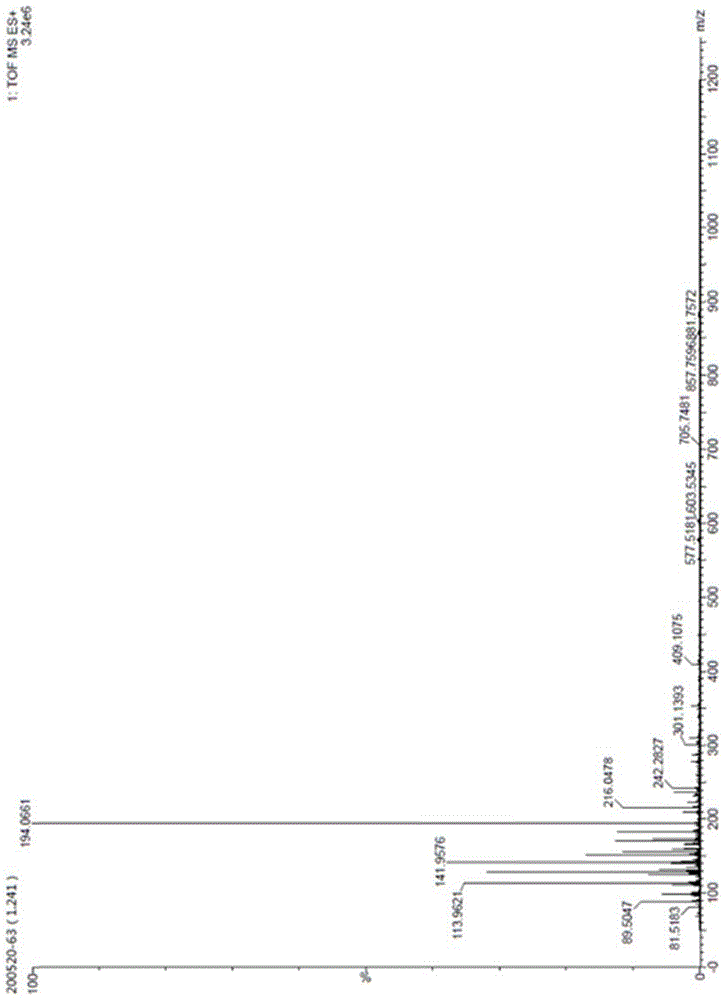
esi-ms(m/z):esi-ms正离子质谱图中在m/z194处的较强离子峰对应于样品的[m+h]+离子,由以上分析可知化合物(ⅲ)的分子量为193,如图1所示。
实施例2:
将50g2,4-二氨基6-羟基-5-甲酰胺基嘧啶(mw169.14,0.30mol)溶于120g回收醋酸(mw60.05,1.66mol)中,缓慢升温至60~70℃,控制温度60~70℃,缓慢滴加85g的含量85%醋酐醋酸溶液(mw102.09,0.71mol),滴加时间控制在7h。
滴毕,开始减压蒸馏回收醋酸溶剂,浓缩干后,得到浓缩残留固体约60.5g即为2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶(ⅱ),粗摩尔收率约95.5%,纯度98.5%。回收醋酸可反复套用。
向化合物(ⅱ)中加入5g催化剂a(四丁基溴化铵)和300g回收二甲苯,开搅拌和加热,升温至115~125,回流脱水反应15h,当分分水器中没有明显水滴产生时,反应结束。开始降温至室温,保温结晶2小时,过滤,干燥得到淡黄色固体粉末约54.1g,即为2-乙酰氨基-6羟基嘌呤(ⅲ),摩尔收率97.9%,纯度98.3%。
实施例3:
将50g(mw193.16,0.26mol)2-乙酰氨基-6羟基嘌呤(ⅲ)和3g醋酸钠溶于500g75%醋酐醋酸溶液中(mw102.09,3.67mol)中,开搅拌和加热,升温至125~135℃,反应8小时。
反应毕,降温至5~15℃,保温结晶2小时,过滤,干燥得到类白色结晶性固体粉末约59.9g,即为n(2),9-二乙酰鸟嘌呤(ⅳ),摩尔收率98.0%,纯度99.1%。过滤的母液检测醋酐含量后可直接套用或蒸馏回收醋酸后套用至步骤(1)。
本实施例制备得到的n(2),9-二乙酰鸟嘌呤结构分析数据如下:
esi-ms(m/z):esi-ms正离子质谱图中在m/z236处的离子峰对应于样品的[m+h]+离子,在m/z194处的较强离子峰对应的9位乙酰基碎片去除后的片段,在m/z152处的离子峰对应的是n上的乙酰基和9位乙酰基碎片去除后的片段,由以上分析可知化合物(ⅳ)的分子量为235,如图2所示。
1h-nmr(400mhz,dmso,ppm):δ:2.21(s,3h,ch3),2.81(s,3h,ch3),8.45(s,1h,ch),11.76(s,1h,nh),12.23(s,1h,oh)如图3所示。
实施例4:
将50g(mw193.16,0.26mol)2-乙酰氨基-6羟基嘌呤(ⅲ)和0.5g醋酸钠(母液套用醋酸钠补加量减少)溶于500g醋酐含量为70%过滤母液中(mw102.09,3.43mol)中,开搅拌和加热,升温至125~135℃,反应10小时。
反应毕,降温至5~15℃,保温结晶2小时,过滤,干燥得到类白色结晶性固体粉末约60.2g,即为n(2),9-二乙酰鸟嘌呤(ⅳ),摩尔收率98.5%,纯度98.8%。过滤的母液检测醋酐含量后可直接套用或蒸馏回收醋酸后套用至2-乙酰氨基-4-氨基-6-羟基-5-甲酰胺基嘧啶的合成中,或者套用至2-乙酰氨基-6羟基嘌呤的合成。
对比例1
直接将50g2,4-二氨基6-羟基-5-甲酰胺基嘧啶加入到186g醋酸酐中进行反应,反应温度控制在60~70℃,反应时间控制在6h。反应完成,减压浓缩干后,得到浓缩残留固体约72.3g,纯度仅为86.7%,二乙酰化杂质约为12.8%。由此可知,步骤(1)中,反应溶剂体系的选择以及滴加方式的选择,对产品纯度具有显著影响。
对比例2
将50g2-乙酰氨基-6羟基嘌呤(ⅲ)置于500g醋酸酐中,加热至125~135℃,反应8小时。反应毕,降温至5~15℃,保温结晶2小时,过滤,干燥得到的产品为淡黄色固体粉末,外观不佳,纯度为96.8%。由此可知,采用混酸体系,并配合催化剂,可以对产品外观和纯度起到决定性作用。
- 还没有人留言评论。精彩留言会获得点赞!