咪唑并噁嗪晶体、含有所述晶体的药物组合物和制备所述晶体的方法与流程
本申请是2016年2月26日提交的发明名称为“咪唑并噁嗪晶体、含有所述晶体的药物组合物和制备所述晶体的方法”、申请号为“201680018450.0”的发明专利申请的分案申请。
本发明涉及可用作抗肿瘤剂的具有优异贮存稳定性的咪唑并噁嗪(imidazooxazine)化合物的新型晶体;包括该晶体的药物组合物以及用于制备该晶体的方法。
背景技术:
通常,当化合物用作药品的活性成分时,化合物的化学稳定性和物理稳定性是必要的,以确保稳定的质量保持和/或容易的贮存管理。因此,稳定型晶体是该化合物的优选最终形式。一般而言,通常选择最稳定型晶体作为药品的活性药物成分。另外,在ich(人用药物注册技术要求国际协调会议)指南中的用于药品残留溶剂的指南规定了各种溶剂的可接受性和可接受量。因为用于制备药品的溶剂通常是有毒的,所以考虑到安全性,优选在制造过程中尽可能地减少残留溶剂。
目前,已经报道了可用作抗肿瘤剂的多种akt抑制剂。专利文献1公开了由下式(1)表示的咪唑并噁嗪化合物(化学名:反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基环丁醇(该化合物可在下文称为“化合物(1)”)),其作为具有优异akt抑制作用和抗肿瘤活性的化合物。
然而,该化合物的晶体形式是完全未知的,并且目前尚未报道用于在确保充分再现性的同时制备高纯度咪唑并噁嗪化合物的稳定型晶体的方法。
引用列表
专利文献
专利文献1:wo2012/137870
技术实现要素:
技术问题
本发明的目的是提供可用作抗肿瘤剂的化合物(1)的晶体,该晶体具有优异稳定性以及关于其制备和制剂方面的有利特征。
问题的解决方案
本发明的发明人进行了广泛研究以解决该问题;并且本发明人发现了晶体ii,其通过在含水乙醇中混悬加热由式(1)表示的咪唑并噁嗪化合物而获得。此外,通过进行进一步将晶体ii混悬在热水中的方法,本发明人成功获得化合物(1)的晶体i从而完成本发明,该晶体具有优异稳定性和在其作为药品的制备和制剂方面的有利特征。
更具体地,从如ich指南等中规定的药品的工业化生产的角度来看,需要各种品质,该品质包括活性药物成分中的杂质量、残留溶剂量以及活性药物成分本身的稳定性等。因此,本发明人一直在考虑化合物(1)的最终晶体形式以便开发作为药品的活性药物成分的化合物。如稍后描述的参考例所示,通过nmr分析确认通过在专利文献1中公开的制备方法获得的化合物(1)的粗产物中的残留乙腈。因为优选在制备药品期间从粗产物中除去乙腈,所以通过将化合物(1)的粗产物在含水乙醇中进行混悬加热来除去残留乙腈。然而,因为在该步骤中获得的晶体ii具有易于含有乙醇的特征,所以制备过程中的残留乙醇可超过由ich指南限制的量。为了解决这个问题,本发明人将所得的晶体ii进一步在热水中混悬,从而成功获得不含残留溶剂的晶体i。因此,本发明人完成了本发明。
更具体地,本发明提供以下内容。
项目1.一种反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基)环丁醇的晶体,所述晶体在粉末x射线衍射光谱中具有至少5个选自7.7°、9.5°、10.3°、12.3°、14.5°、15.6°、16.3°、17.8°、18.3°、19.3°、20.9°、22.8°、24.2°、25.7°、26.8°、27.7°、29.0°和30.1°的衍射角(2θ±0.1°)的峰。
项目2.根据项目1所述的晶体,其中所述晶体在粉末x射线衍射光谱中具有在7.7°、9.5°、10.3°、12.3°、14.5°、15.6°、16.3°、17.8°、18.3°、19.3°、20.9°、22.8°、24.2°、25.7°、26.8°、27.7°、29.0°和30.1°处的衍射角(2θ±0.1°)的峰。
项目3.根据项目1或项目2所述的晶体,其中所述晶体在差示扫描量热法(dsc)中具有在230℃附近的吸热峰。
项目4.一种药物组合物,其包括根据项目1至项目3中任一项所述的晶体。
项目5.一种用于口服的药物组合物,其包括根据项目1至项目3中任一项所述的晶体。
项目6.一种用于制备根据项目1至项目3中任一项所述的晶体的方法,所述方法包括以下步骤:
(1)将反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基)环丁醇在含水乙醇中混悬加热以获得混悬液;
(2)从步骤(1)获得的所述混悬液获得反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基)环丁醇的固体;以及
(3)将步骤(2)获得的反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基)环丁醇的固体在热水中混悬加热。
本发明的有益效果
因为本发明的咪唑并噁嗪化合物的晶体i具有优异的贮存稳定性,所以该晶体i在例如纯度、处理(较低吸湿性)、流动性、可磨性和/或质量控制等方面优于其它晶体形式,并且因此该晶体可用作适用于药品的药物制剂的晶体。
另外,本发明的晶体i含有不超过由ich中针对药品中残留溶剂的指南所限制的标准值的量的残留溶剂,并且因此作为药品是安全的。
附图说明
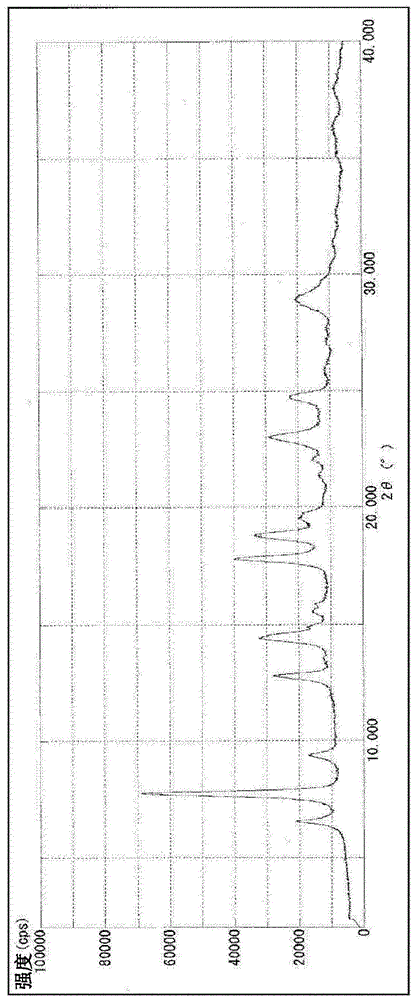
图1示出在实施例1中获得的化合物(1)的晶体ii的粉末x射线衍射光谱(纵轴表示强度(cps),而横轴表示衍射角(2θ))。
图2示出在实施例1中获得的化合物(1)的晶体ii的差示扫描量热(dsc)曲线。
图3示出在实施例1中获得的化合物(1)的晶体i的粉末x射线衍射光谱(纵轴表示强度(cps),而横轴表示衍射角(2θ))。
图4示出在实施例1中获得的化合物(1)的晶体i的差示扫描量热(dsc)曲线。
图5示出在稳定性测试中分析类似物的增加量的hplc数据。
具体实施方式
在本说明书中,术语“化合物(1)”是指“反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基)环丁醇”,并且包括无定形和晶体两者。
在本说明书中,术语“晶体”和“无定形”根据一般定义来解释。
由于粉末x射线衍射本身的特征,衍射角和整个峰图案在确定晶体的同一性方面是重要的。因为粉末x射线衍射图案的相对强度根据晶体生长方向、颗粒尺寸和测量条件而在一定程度上变化,所以不应严格地解释相对强度。
根据各种粉末x射线衍射图案获得的值可根据晶体生长方向、颗粒尺寸、测量条件等而在一定程度上含有误差。因此,在本说明书中,粉末x射线衍射图案中的衍射角(2θ)的值可含有在约±0.1°的范围内的测量误差。
另外,关于差示扫描量热(dsc)曲线中的吸热峰,测量温度可根据每分钟的升温范围、样品的纯度等变化。在本说明书中,术语“附近”意指±5.0℃的范围。
化合物(1)的晶体i优选示出图3所示的粉末x射线衍射光谱和/或图4所示的差示扫描量热(dsc)曲线。
化合物(1)的晶体i的粉末x射线衍射光谱中衍射角(2θ±0.1°)的特征峰包括7.7°、9.5°、10.3°、12.3°、14.5°、15.6°、16.3°、17.8°、18.3°、19.3°、20.9°、22.8°、24.2°、25.7°、26.8°、27.7°、29.0°和30.1°。
本发明的化合物(1)的晶体i是示出以上峰的至少5个峰、优选至少8个峰、特别优选所有峰的晶体。
另外,化合物(1)的晶体i的差示扫描量热(dsc)曲线中的吸热测量温度在228℃至232℃附近,优选在230℃附近。
化合物(1)可根据诸如下述参考例的方法进行合成。然而,化合物(1)的合成方法不限于下述参考例。
本发明的化合物(1)的晶体i可通过诸如以下的方法制备,该方法包括:将由式(1)表示的咪唑并噁嗪化合物在含水乙醇中混悬加热,之后在热水中混悬加热。
更具体地,将化合物(1)混悬在量为1倍至100倍、优选地1倍至50倍的乙醇与量为1倍至100倍、优选地1倍至50倍的水的混合物中。在搅拌下将混合物加热1小时至100小时、优选1小时至24小时,然后冷却,通过过滤收集并干燥,从而得到晶体ii。可以适当选择水和乙醇的混合物的混合比率;因此混合比率优选使得水:乙醇(v/v)的比率为1:0.1~10,更优选地1:0.5~2,且特别优选地1:1。该步骤的加热温度可适当设定;因此加热温度优选为40℃或更高,并且更优选为40℃至60℃。
随后,将所得的晶体ii混悬在量为1至100倍、优选10倍至50倍的水中,并且将混悬液在回流下加热1小时至120小时并且优选12小时至48小时,然后冷却,通过过滤收集并且干燥,从而获得白色粉末的晶体i。该步骤中的加热温度可适当设定;因此,加热温度优选为70℃或更高,并且更优选为70℃至90℃。
为了加速晶体i的结晶,可添加适量的化合物(1)的晶体i或含有晶体i的混合晶体作为晶种。基于溶剂的量,待添加晶种的量为0.01~5(w/v)%,优选0.03~1(w/v)%。另外,可在搅拌下执行结晶,以便缩短结晶时间并控制粒径。
在本发明中,术语“冷却”意指将溶液的温度保持在40℃或更低,优选在室温或更低。冷却时间优选为0.5小时或更长,并且更优选为1小时或更长。
析出的晶体可通过已知分离和纯化手段(诸如过滤、用水洗涤或在减压下干燥)从溶解有晶体的溶液、混合溶液等中分离和纯化。
本发明的化合物(1)的晶体i示出优异固体稳定性(贮存稳定性等)。在开发药品时,重要的是候选化合物在工业操作和质量保持方面均示出固体稳定性。另外,本发明的化合物(1)的晶体i中的残留溶剂的量不超过由ich指南限制的量。在药品的安全性方面,非常重要的是未观察到残留溶剂。因此,本发明的化合物(1)的晶体i示出作为药品或药品的活性药物成分的优异性质。
本发明的化合物(1)的晶体示出优异akt抑制活性,可用作治疗例如癌症和肿瘤的药剂。另外,本发明的化合物(1)的晶体示出对于akt的显著优异的选择性,并且具有这样的优点:由其他激酶的抑制导致的副作用较小。
本发明的化合物(1)的晶体示出优异的akt抑制活性。在本说明书中,“akt”包括人akt和非人哺乳动物akt,并且优选人akt。此外,“akt”包括其同种型。
另外,由于其优异的akt抑制活性,本发明的化合物(1)的晶体可用作预防或治疗akt相关疾病诸癌症的药剂。
在将本发明的化合物(1)的晶体用于预防和治疗疾病的药剂时,可在粉碎之后或不粉碎的情况下根据预防或治疗目的以各种剂型使用该晶体。剂型的实例包括口服制剂诸如片剂、胶囊、颗粒、细粒、粉剂或干糖浆;栓剂、吸入剂、滴鼻剂、软膏、贴剂和注射剂。在这些之中,优选口服制剂。这些药物组合物可通过本领域技术人员已知的常规制备方法使用药学上可接受的载体来制备。
作为药物载体,可使用通常用作制剂材料的各种有机或无机载体材料作为固体制剂中的赋形剂、粘合剂、崩解剂、润滑剂或着色剂;或作为液体制剂中的溶剂、增溶剂、混悬剂、等渗剂、缓冲剂或舒缓剂。另外,如果需要,还可使用药物制剂添加剂,诸如防腐剂、抗氧化剂、着色剂、甜味剂和稳定剂。
口服固体制剂可如下制备。使用常规的方法,将赋形剂、任选地与其它赋形剂、粘合剂、崩解剂、润滑剂、着色剂、掩味或调味剂等一起添加到本发明的化合物中,以制备片剂、包衣片剂、颗粒、粉剂、胶囊等。
赋形剂的实例包括乳糖、蔗糖、d-甘露糖醇、葡萄糖、淀粉、碳酸钙、高岭土、微晶纤维素和硅酸酐。
粘合剂的实例包括水、乙醇、1-丙醇、2-丙醇、单糖浆、葡萄糖溶液、α-淀粉溶液、明胶溶液、d-甘露糖醇、羧甲基纤维素、羟丙基纤维素、羟丙基淀粉、甲基纤维素、乙基纤维素、虫胶、磷酸钙和聚乙烯吡咯烷酮。
崩解剂的实例包括干淀粉、藻酸钠、琼脂粉、碳酸氢钠、碳酸钙、月桂基硫酸钠、硬脂酸单甘油酯和乳糖。
润滑剂的实例包括经纯化滑石、硬脂酸钠、硬脂酸镁、硼砂和聚乙二醇。
着色剂的实例包括氧化钛和氧化铁。
掩味或调味剂的实例包括蔗糖、野橙皮、柠檬酸和酒石酸。
栓剂如下制备。将本领域已知的药物载体诸如聚乙二醇、羊毛脂、可可脂和脂肪酸甘油三酯添加到本发明的化合物中,任选地与吐温80
软膏如下制备。根据要求将常规碱、稳定剂、润湿剂、防腐剂等添加到本发明的化合物中,并且使用常规方法混合并配制成药物。碱的实例包括液体石蜡、白凡士林、白蜂蜡、辛基十二烷醇和石蜡。防腐剂的实例包括对羟基苯甲酸甲酯、对羟基苯甲酸乙酯和对羟基苯甲酸丙酯。
可通过使用常规方法用以上软膏、霜膏、凝胶、糊剂等涂布普通支承物来制备贴剂。支承物的实例包括由棉、短纤维和化学纤维制成的织物或无纺织物;以及软质聚氯乙烯、聚乙烯、聚氨酯等的膜和泡沫片材。
注射剂如下制备。将ph调节剂、缓冲剂、稳定剂、等渗剂、局部麻醉剂等添加到本发明的化合物中,以使用常规方法制备皮下注射剂、肌内注射剂或静脉内注射剂。在这种情况下的可用ph调节剂和缓冲剂的实例包括柠檬酸钠、乙酸钠和磷酸钠。可用稳定剂的实例包括焦亚硫酸钠、edta、巯基乙酸和硫代乳酸。可用的局部麻醉剂的实例包括盐酸普鲁卡因和盐酸利多卡因。可用的等渗剂的实例包括氯化钠、葡萄糖、d-甘露糖醇和甘油。
待引入到各个这样的剂量单位形式中的化合物(1)的晶体i的量取决于被施用化合物的患者的病症、该剂量单位形式的剂型等。然而,通常,在口服剂的情况下,化合物(1)的晶体i的量为每剂量单位形式约0.05~1000mg。在注射剂的情况下,化合物(1)的晶体i的量为每剂量单位形式0.1~500mg;且在栓剂或外用制剂的情况下,化合物(1)的晶体i的量为每剂量单位形式约1~1000mg。
另外,在此类剂型中的药物的日剂量取决于患者的病症、体重、年龄、性别等,并且不可一般化。然而,例如,成人(体重:50kg)的日剂量通常可为约0.05~5000mg,且优选0.1~1000mg;并且优选以每天一个剂量、或每天两至三次的分剂量施用。
另外,如上所述,可例如通过以下方式获得晶体i:将由式(1)表示的咪唑并噁嗪化合物在含水乙醇中混悬加热以获得晶体ii,然后将晶体ii混悬在热水中。因此,在本发明中,化合物(1)的晶体ii也可用作制备晶体i的中间体。因此,本发明还提供化合物(1)的晶体ii。
化合物(1)的晶体ii在粉末x射线衍射光谱中的衍射角(2θ±0.1°)的特征峰包括6.6°、7.8°、9.3°、12.8°、13.6°、14.4°、17.8°、18.8°、19.5°、21.4°、22.5°、23.0°、24.8°、27.8°和29.0°。
本发明的化合物(1)的晶体ii为具有以上峰的至少5个峰、优选至少8个峰、特别优选全部峰的晶体。
实施例
下面参考实施例更具体地解释本发明;然而,本发明不限于这些实施例。下面通过实施例充分描述本发明;然而,应当理解,由本领域技术人员作出的各种改变和修改是可能的。因此,只要此类改变和修改不脱离本发明的范围,那么此类改变和修改就包括在本发明中。
在实施例中,除非另有说明,否则使用市售的试剂。通过使用al400(400mhz,由jeol制造)测量nmr光谱。当氘代溶剂含有四甲基硅烷时,使用四甲基硅烷作为内标。在其他情况下,使用保留在nmr溶剂中的非氘代质子的峰作为内标进行测量。所有δ值均以ppm表示。
每个符号代表以下内容。
s:单峰
d:双峰
t:三峰
dd:双双峰
m:多重峰
brs:宽单峰
dmso-d6:氘代二甲基亚砜
cdcl3:氘代氯仿
粉末x射线衍射测量
粉末x射线衍射是通过以下方式执行:根据需要使用玛瑙研钵轻轻粉碎适量的测试物质,并根据以下测试条件测量测试物质。
设备:miniflexii,rigakucorporation
靶标:cu
x射线输出功率:15ma、30kv
扫描范围:2.0°至40.0°
步长大小:0.010°
扫描速度:5.00℃/min。
发散狭缝:1.25°
散射狭缝:打开
接收狭缝:打开
根据指定用于每个设备的方法和程序执行设备的处理,包括数据处理。
根据各种光谱获得的值可根据晶体生长方向、颗粒尺寸、测量条件等而在一定程度上变化。因此,这些值不应该被严格的解释。
热分析测量(差示扫描量热法(dsc测量))
根据以下测试条件执行dsc测量。
设备:ta仪器q1000
样品:约1mg
样品容器:铝容器
升温速度:以5℃/min升至250℃
气氛:氮气
氮气流速:50ml/min
根据指定用于每个设备的方法和程序执行设备的处理,包括数据处理。
参考例1:反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基环丁醇(化合物(1))的合成
将根据wo2012/137870中公开的方法合成的(反式-3-羟基-3-甲基-1-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基)环丁基)氨基甲酸叔丁酯(27.3g)混悬在乙腈(550ml)中,并且在室温下添加甲磺酸(22ml)。在室温下搅拌1小时后,加入1m氢氧化钠水溶液(340ml)以淬灭反应。因为反应混合物分离成两层,所以加入水(340ml)以获得均质溶液。在55℃下减压蒸馏掉830ml溶剂。将其恢复到室温后,通过过滤收集所生成的固体。通过过滤收集的固体用水洗涤,并且在70℃下干燥,从而获得标题化合物(化合物(1))的粗产物(16.7g),其为浅黄色固体。
1h-nmr(cdcl3):δ9.26(1h,s),8.49(1h,d,j=5.6hz),7.57-7.52(2h,m),7.52-7.43(3h,m),7.39-7.32(2h,m),7.28-7.21(2h,m),6.99(1h,dd,j=5.6,0.5hz),5.76(2h,s),2.65-2.57(2h,m),2.39-2.31(2h,m),1.64(3h,s)。
残留乙腈峰:δ2.01(3h,s)
由nmr峰面积计算的残留乙腈量为约600ppm。
实施例1:反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基环丁醇(化合物(1))的晶体ii的制备
将在参考例1中获得的化合物(1)的粗产物(480mg)混悬于乙醇(2ml)和水(2ml)的混合溶剂中。将混悬液在50℃下搅拌3小时,并且然后冷却至室温。通过过滤收集固体,用水洗涤,并在40℃下干燥,从而得到标题化合物的晶体ii(438mg),其为白色固体。
1h-nmr(dmso-d6):δ9.01(1h,d,j=0.5hz),8.48(1h,d,j=5.4hz),7.56-7.46(3h,m),7.45-7.37(4h,m),7.32-7.28(2h,m),7.21(1h,dd,j=5.4,0.5hz),5.93(2h,s),4.74(1h,s),3.32(1h,s),2.39-2.28(2h,m),2.18-2.07(2h,m),1.95(2h,brs),1.49(3h,s)。
残留乙醇峰:δ4.34(1h,m)、3.43(2h,m)、1.04(3h,t,j=7.3hz)。
由nmr峰面积计算的残留乙醇量为约14000ppm。
图1示出粉末x射线衍射光谱。特征衍射角(2θ±0.1°):6.6°、7.8°、9.3°、12.8°、13.6°、14.4°、17.8°、18.8°、19.5°、21.4°、22.5°、23.0°、24.8°、27.8°和29.0°。
图2示出差示扫描量热(dsc)曲线。
差示扫描量热(dsc)曲线中的吸热峰:228℃至232℃附近。
反式-3-氨基-1-甲基-3-(4-(3-苯基-5h-咪唑并[1,2-c]吡啶并[3,4-e][1,3]噁嗪-2-基)苯基环丁醇(化合物(1))的晶体i的制备
将以上获得的晶体ii(16.16g)混悬于水(160ml)中,并且在80℃下加热的同时搅拌31小时,然后冷却至室温。通过过滤收集固体,用水洗涤,并在80℃下干燥,从而得到标题化合物的晶体i(15.98g),其为白色固体。
1h-nmr(cdcl3):δ9.26(1h,s),8.49(1h,d,j=5.6hz),7.57-7.52(2h,m),7.52-7.43(3h,m),7.39-7.32(2h,m),7.28-7.21(2h,m),6.99(1h,dd,j=5.6,0.5hz),5.76(2h,s),2.65-2.57(2h,m),2.39-2.31(2h,m),1.64(3h,s)。
未观察到残留溶剂峰。
图3示出粉末x射线衍射光谱。特征衍射角(2θ±0.1°):7.7°、9.5°、10.3°、12.3°、14.5°、15.6°、16.3°、17.8°、18.3°、19.3°、20.9°、22.8°、24.2°、25.7°、26.8°、27.7°、29.0°和30.1°。
图4示出差示扫描量热(dsc)曲线。
差示扫描量热(dsc)曲线中的吸热峰:228℃至232℃附近。
实施例2:固体稳定性测试
在40℃贮存6个月后,测试根据实施例1获得的咪唑并噁嗪化合物的晶体i的贮存稳定性。
贮存条件:40℃(气密)
测量点:第六个月
量:约30mg
容器:棕色玻璃瓶
使用上述方法测量贮存后的样品的粉末x射线衍射。
通过hplc根据以下方式分析类似物量的变化。
制备样品溶液的方法:将样品溶解在50%乙腈中,使得样品浓度变为0.5mg/ml。
柱:acentisexpressc18,4.6×150mm,2.7μm,
柱温:40℃
柱流速:1.5ml/min
流动相:a;0.1%磷酸盐,b;乙腈
检测uv:210nm
梯度:
结果显示出,晶体i中未观察到粉末x射线衍射图案的改变,并且因此晶体i是非常稳定的晶体。另外,晶体i仅含有少量类似物,并且在40℃下贮存6个月后,类似物的量不增加。
- 还没有人留言评论。精彩留言会获得点赞!