一种新的I型CRISPR/Cas系统及其应用
一种新的i型crispr/cas系统及其应用
技术领域
1.本发明涉及生物工程领域,具体而言,本发明涉及一种新的基于crispr/cas9系统的基因组编辑系统及其应用。
背景技术:
2.crispr(clustered regularly interspaced short palindromic repeats,crispr)系统是细菌和古细菌抵御外源dna或rna入侵的一种适应性免疫系统。在该系统基础上发展的基因组编辑工具crispr/cas系统目前已广泛应用于植物、动物和微生物的基因组编辑。cas核酸酶在向导rna的靶向作用下对dna双链进行切割,之后通过dna修复途径进行修复。大多数细菌不具有dna双链断裂末端修复的非同源末端连接(nhej)途径,可向细菌中转入同源片段通过同源重组(hr)进行dna修复,实现基因组编辑。
3.在自然界中,将近一半的细菌自身就拥有crispr/cas系统,利用细菌自身的crispr/cas系统来编辑基因组可以消除表达外源cas蛋白对细菌的毒性作用,并且,由于不需要向细菌中转化表达cas核酸酶的质粒,可极大的简化基因编辑系统。目前,已在细菌clostridium、lactobacillus和pseudomonas的一些菌株中,通过转入带有同源修复模板并表达靶定基因组的crispr阵列的质粒,成功利用自身的i型crispr/cas系统实现了基因编辑。目前在植物病原菌中通过内源的crispr/cas系统进行基因组编辑的方法还未有报道。
技术实现要素:
4.本技术的发明人在植物病原菌水稻黄单胞菌中鉴定到一个新的i型crispr/cas系统,并实现了利用内源crispr/cas系统进行高效的基因组编辑。本发明可实现无标记基因残留的高效快速的基因组编辑方式。本发明提供了构建的基于内源crispr/cas系统进行基因组编辑的表达载体。本发明还提供用所述基因组编辑系统编辑细胞基因组的方法。
5.具体而言,发明人通过如下项目所公开的技术方案解决了本领域中存在的上述问题:
6.1.一种基于植物病原菌内源的crispr/cas系统的基因组编辑系统,其包含构建于一个或多个载体中的可操作地连接的λ-red同源重组系统的编码核酸序列、用于靶定到基因组特定位置的crrna的编码核酸序列和用于目标突变体构建的同源重组片段的核酸序列。
7.2.项目2所述的基因组编辑系统,其中所述载体为质粒。
8.3.项目1或2所述的基因组编辑系统,其中所述λ-red同源重组系统的编码核酸序列构建于一个载体上,并且用于靶定到基因组特定位置的crrna的编码核酸序列和用于目标突变体构建的同源重组片段的核酸序列构建于另一个载体上。
9.4.项目1-3任一项所述的方法,其中所述crrna的加工成熟的核苷酸序列如seq id no:28-seq id no:32任一项所示。
10.5.对细胞进行基因编辑的方法,其包括将项目1-6任一项所述的基因组编辑系统
引入所述细胞,其中所述细胞为细菌,优选黄单胞菌属(xanthomonas),更优选水稻黄单胞菌(xanthomonas oryzae)的细胞,其中所述的基因组编辑系统和所述细胞内源的crispr/cas组合实现对细胞基因组的编辑。
11.6.项目1-6任一项所述的基因组编辑系统在制备基因编辑细胞中的用途,其中所述基因编辑为基因敲除及替换等,优选多个基因的编辑。
12.7.一种分离的crispr/cas系统,其包含或组成为如seq id no:10所示的核苷酸序列。
13.在另一个方面,本发明对内源crispr/cas系统进行了详细分析,确定了crrna的加工过程及方式、识别的pam序列。
14.在另一个方面,本发明对细菌中的质粒进行高效的编辑,可进行质粒的消除,也可对质粒进行同源重组编辑。
15.在另一个方面,本发明涉及对crrna与dna间的错配对编辑效率的影响进行了分析。
附图说明
16.图1:pxo99a内源的i-c型crispr系统特征示意图。其中图1a显示pxo99内源的i-c型crispr/cas系统组成。图1b为前体crrna(precursor crispr rna,pre-crrna)加工成成熟crrna示意图。黑色箭头代表切割位点。一个成熟的crrna包含5’端11nt的柄,33-37nt的间隔序列以及3’端20nt的颈环结构。图1c为pxo99内源的crispr/cas系统所识别的pam预测结果。
17.图2:内源crispr/cas系统可实现对pxo99a中质粒的编辑。其中图2a为测试内源crispr/cas系统所用质粒图谱,其中pseva-egfpt表达可靶定到egfp上的crrna,pseva-egfptd表达可靶定到egfp上的crrna并带有可与egfp基因发生同源重组的同源片段;图2b为转化pseva-egfpt后的结果示意图,由于内源crispr/cas系统对质粒dna的切割,使得转化后的细菌丧失壮观霉素抗性;图2c为转化pseva-egfptd后的结果示意图,由于利用同源片段对切割的质粒进行修复,转化后的细菌带有壮观霉素抗性;图2d显示带有不同组分的pseva系列载体转入含有phm1-egfp的pxo99a的菌落数统计结果及相关图片,转化用的质粒dna量一致,转化后的细菌涂在含相同浓度抗生素的平板上,标尺=1cm,cfu为菌落形成单位,ha表示同源臂。
18.图3:内源crispr/cas系统可实现对xanb2基因的切割和编辑。其中图3a显示设计的靶向xanb2的间隔序列,pam用下划线标注。图3b显示pxo99内源i-c型crispr/cas系统与不同组分组合的突变效率,转化用的质粒dna量一致,转化后的细菌涂在含相同浓度抗生素的平板上,ha为同源臂。
19.图4:错配对pxo99内源的crispr/cas系统的编辑效率的影响。其中图4a显示设计的crrna的突变类型,突变位点用下划线标注。图4b为不同突变类型的crrna的突变效率,对照为未突变的crrna,转化用的质粒dna量一致,转化后的细菌涂在含相同浓度抗生素的平板上。
具体实施方式
20.为使本发明的目的、技术方案和优点更加清楚明白,以下结合具体实施例,并参照附图,对本发明作进一步的详细说明。
21.除非另外说明,否则本文所用的百分比浓度为质量体积百分比浓度。
22.除非另外说明,否则本文中所用的术语具有本领域所通常理解的含义。
23.特别地,本文所用的术语“可操作地连接”是指例如载体(例如质粒)中所连接的序列以实现其功能的方式连接。
24.方法
25.phm1-egfp质粒的构建
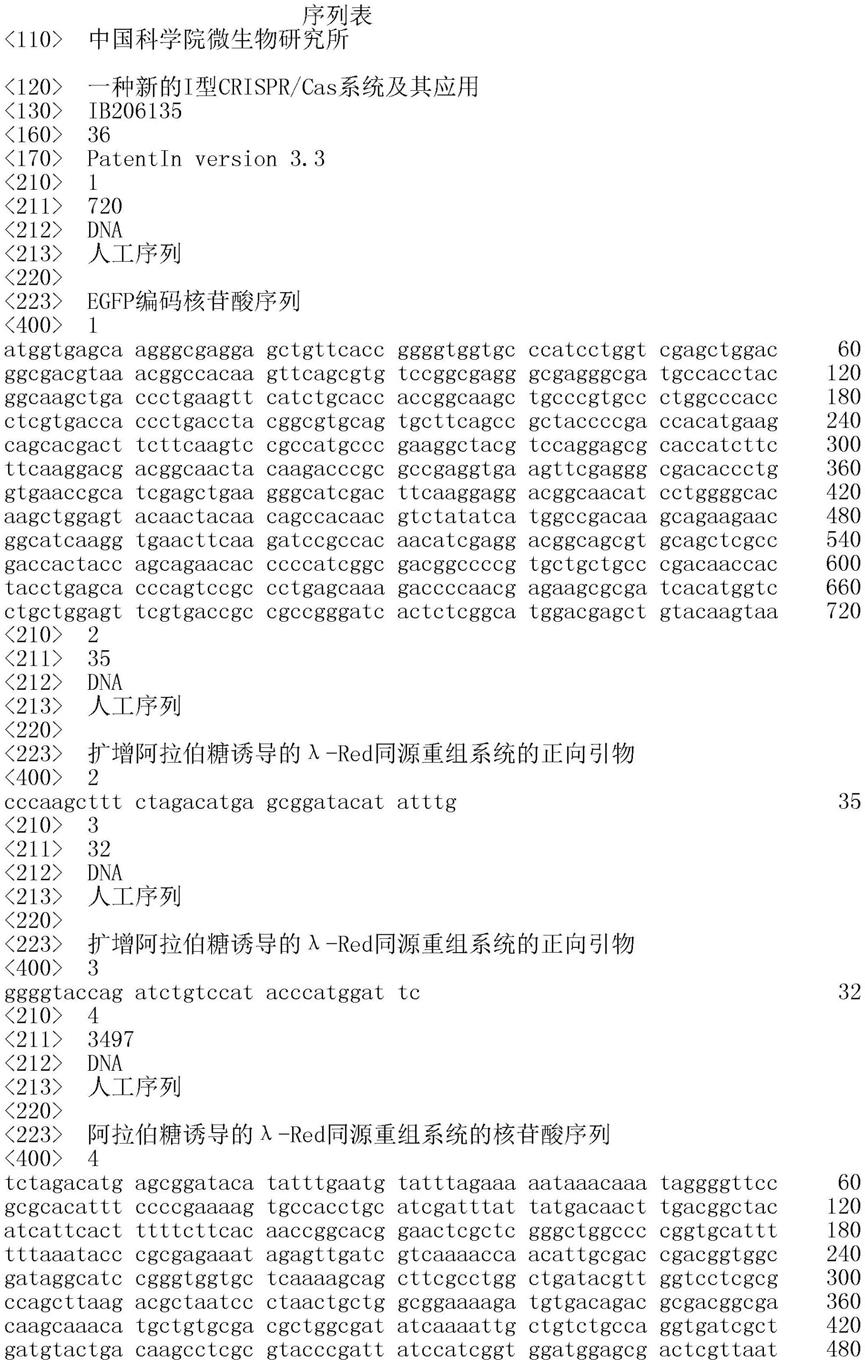
26.用psti和ecori两种限制性内切酶对质粒pegfp(购自clontech公司)进行消化,获得egfp片段(egfp的编码核酸序列见seq id no:1),然后将egfp片段与经psti和ecori两种限制性内切酶酶切的phm1载体(本实验室保存质粒,genbank:ef059993.1)通过t4 dna连接酶(thermo公司,货号el0011)连接,转化dh5α。通过设计的phm1载体的通用引物(5
’-
tgcgatgaatgatttcaaactag-3’,seq id no:33;5
’-
cagagaatgaggaacaccagatg-3’,seq id no:34)进行菌落pcr后挑选阳性菌落测序鉴定,25μl反应体系(试剂购自tiangen公司)如下:10xtaq反应缓冲液2.5μl;10mm dntps 2μl;10μm正向引物1.25μl;10μm反向引物1.25μl;taq dnapolymerase 0.25μl;加水至25μl后加入菌落模板。反应程序为:94℃5min;94℃30s,60℃30s,72℃1min,30个循环;72℃5min。
27.pxo99a电击感受态的制备及电击转化方法
28.(1)在m210固体平板上划线活化水稻黄单胞菌白叶枯致病变种菌株pxo99a(本实验室保存菌株,ncbi reference sequence:nc_010717.2),28℃培养2-3天后接种至5ml液体m210培养基中,28℃、220rpm培养过夜。
29.(2)将过夜培养的菌液按照1.5%-2%(体积比)的比例转接至250ml的m210液体培养基(5g蔗糖,8g酶水解酪素,4g酵母提取物,3g k2hpo4,0.3g mgso4.7h2o,定容至1l,ph值为7.0)中,28℃、220rpm培养至od
600
值在0.5-0.6之间。
30.(3)将菌液至于冰上冷却20min,然后将菌液转移至离心筒中于4℃、4000rpm离心回收菌体,并用预冷的10%的甘油洗涤菌体3次。最后用1ml预冷的10%的甘油重悬菌体,每100μl分装于一个1.5ml离心管中,液氮速冻后放入-80℃冻存备用。
31.(4)将目的质粒加入解冻的pxo99a感受态,然后转移至预冷的电击杯中,用bio-rad micropulser电击仪在agr参数项下进行电击,电击后加入900μl ps液体培养基(10g蛋白胨,10g蔗糖,1.27g l-谷氨酸钠
·
h2o,定容至1l,ph值为7.0)于28℃摇床复苏,4小时后将菌液涂至含有相应抗生素的psa平板上,放于28℃温箱中培养2-3天。
32.phm1-λ-red质粒的构建
33.以pcas-rk2t载体(molecularcloud公司,产品编号mc_0000261)为模板,以设计的5
’-
cccaagctttctagacatgagcggatacatatttg-3’(正向引物,seq id no:2)和5
’-
ggggtaccagatctgtccatacccatggattc-3’(反向引物,seq id no:3)为引物进行pcr扩增λ-red片段,片段长度约3500bp(核苷酸序列见seq id no:4)。使用q5高保真聚合酶(neb公司,货号:m0491l)扩增插入片段,扩增体系为50μl:5xq5反应缓冲液10μl;10mm dntps1μl;10μm正向引物2.5μl;10μm反向引物2.5μl;q5 high-fidelity dnapolymerase 0.5μl;dna模板1ng;
加水至50μl。反应程序为:98℃30s;98℃10s,60℃20s,72℃30s,30个循环,72℃2min。之后对产物进行纯化。通过hindiii和kpni两种限制性内切酶对纯化后的pcr产物进行酶切,然后将酶切后的片段与经hindiii和kpni酶切的phm1载体连接,转化dh5α,菌落pcr后挑选阳性菌落测序鉴定。
34.表达crrna的pseva-ic-crrna载体的构建
35.(1)改造质粒pseva-gric6t(molecular cloud公司,产品编号mc_0000262),构建pseva-ic-crrna。改造分两个方面,一是将sgrna序列替换为内源i-c型crispr阵列。两个31bp的重复序列间为含bsai酶切位点的序列(5
’-
tgagacctcgctgggtctca-3’),便于crrna spacer的构建。操作步骤如下:以质粒pseva-gric6t(molecular cloud公司,产品编号mc 0000262)为模板,进行两轮pcr扩增,第一轮引物5
’-
ctcacgggcgcgtggattgaaacgtgctttttttgaggagctcggtac-3’(正向引物,seq id no:5)和5
’-
gtgaggacgcgacgctagcattatacctaggactgagctag-3’(反向引物,seq id no:6),之后以扩增产物为模板,进行第二轮扩增,引物为5
’-
gtgggtctccgtcgcgtcctcacgggcgcgtggattgaaac-3’(正向引物,seq id no:7)和5
’-
agaggtctctgtttcaatccacgcgcccgtgaggacgcgacgctagcattatac-3’(反向引物,seq id no:8),之后对产物进行纯化。通过takara mutanbest kit试剂盒按照使用说明书对产物片段进行环化后,转化大肠杆菌dh5α感受态细胞。菌落pcr和测序鉴定获得pseva-ic-crrna载体。二是将质粒pseva-gric6t上的同源片段换成多克隆位点(5
’-
ggatcccgtcgactctgcaggcatgcaagctt-3’),最终质粒共包含7个单酶切位点,便于同源片段的连接。pseva-ic-crrna的相关的核苷酸序列如seq id no:9所示。
36.(2)根据pam设计靶定到特定基因的间隔序列,并在ncbi上进行比对,确保不会产生脱靶。
37.(3)在设计的间隔序列正向引物5’端加上aaac,反向引物5’端加上cgac。引物合成退火后与bsai酶切消化过的pseva-ic-crrna进行连接,即得可表达靶定到x基因位点的crrna的质粒pseva-ic-xt。
38.(4)在要敲除或编辑的基因上下游选择合适的区域作为上下游的同源臂。通过q5高保真聚合酶从pxo99a基因组扩增上下游同源臂,之后通过融合pcr将两个同源臂融合成一个片段,通过酶切连接的方式将同源片段构建到质粒pseva-ic-xt上,得到质粒pseva-ic-xtd。
39.内源crispr/cas系统在pxo99a中应用的基因编辑方法
40.(1)将phm1-λ-red载体转入pxo99a。
41.(2)制备含有质粒phm1-λ-red的pxo99a电击感受体细胞。在转接培养至od
600
值在0.2-0.3时,加入10mm的l-阿拉伯糖诱导λ-red重组系统表达。继续培养至od
600
值在0.5-0.6时,制备电击感受态细胞。
42.(3)将2μg质粒pseva-ic-xtd电转至上述感受态细胞中,电击后加入900μl ps液体培养基于28℃摇床复苏,4小时后将菌液涂至含有相应抗生素的psa平板上,放于28℃温箱培养2-3天。
43.(4)通过突变体鉴定引物对细菌基因组进行pcr扩增确定是否发生相应的突变,并通过测序验证。
44.实施例1.pxo99a中crispr/cas系统类型分析
45.首先,将pxo99a的基因组序列输入到crisprone网站(https://omics.informatics.indiana.edu/crisprone/),初步预测其基因组含有一套内源的crispr系统,包括7个cas基因和串联的crispr阵列序列,见图1a,相关的核苷酸序列如seq id no:10所示。对cas基因的分析发现,cascade复合物由cas5d、cas8c、和cas7亚基组成,指示pxo99a的cas操纵子是典型的i-c型crispr/cas系统。将基因组序列输入到crisprcasfinder网站(https://crisprcas.i2bc.paris-saclay.fr/crisprcasfinder/index),获得crispr阵列序列的详细统计信息,由保守的31bp的重复序列(5
’-
gtcgcgtcctcacgggcgcgtggattgaaac-3’,seq id no:11)和75个33-37bp的不同的间隔序列组成。
46.如图1b所示,cas5d蛋白进行前体crispr rna(pre-crrna)的加工,最终成熟的crrna由11nt的5’柄端结构、33-37nt的间隔序列和20nt的3’茎环结构组成,完整序列为tggattgaaacn
33-37
gtcgcgtcctcacgggcgcg(分别如seq id no:28-seq id no:32所示,其中n为a、t、c或g任一种,n
33-37
代表33-37个核苷酸)。crrna的3’端茎环结构可以终止cas7亚基沿着crrna的寡聚化,大约7个cas7单体会结合在crrna上,形成cascade复合体的骨架。cas8c亚基行使pam的识别、双链解旋、r-loop的形成和稳定功能。cascade/i-c复合体指导下的稳定的r-loop形成和构象改变,会招募cas3核酸酶/解旋酶靶向目标dna,对dna进行切割。
47.pam序列对于crispr-cas系统功能的行使是必需的。为了鉴定pam序列,将crispr阵列输入crispr target网站(http://bioanalysis.otago.ac.nz/crisprtarget/crispr_analysis.html),以预测其原本抵御的外源dna元件,如噬菌体、质粒、转座元件等。发现靶向序列展示出与噬菌体序列和一部分质粒高度的同源性,证明该内源crispr-cas系统在适应性免疫活动中发挥了实际作用。之后将靶向序列输入weblogo网站(http://weblogo.berkeley.edu/logo.cgi),如图1c所示,分析结果表明该内源系统对5
’-
ttc序列有最大偏好性,其次是5
’-
ttt。这跟之前在bacillus halodurans和侵肺军团菌(legionella pneumophila)里的研究结果相一致。
48.实施例2.利用内源的crispr/cas系统对质粒进行编辑
49.在质粒pseva-gric6t的基础上改造并构建质粒pseva-ic-crrna用于表达crrna。首先利用转有质粒phm1-egfp的pxo99
aegfp
菌株检测pxo99a中crispr/cas系统的dna结合及切割活性。设计一个可靶定到基因egfp的34bp的间隔序列,载体构建步骤如下:将带有末端接头的引物5
’-
aaacatctgcaccaccggcaagctgcccgtgccctggc-3’(seq id no:12)和5
’-
cgacgccagggcacgggcagcttgccggtggtgcagat-3’(seq id no:13)进行梯度退火,后连接到bsai酶切后的质粒pseva-ic-crrna上,得到质粒pseva-ic-egfpt。相关质粒图谱见图2a。同时构建载体pseva-ic-ncrrna,表达不能靶定到基因组及质粒phm1-egfp上的ncrrna作为对照(构建引物见seq id no:14和seq id no:15)。
50.将构建好的两个pseva-ic质粒分别转入pxo99
aegfp
菌株中,pseva-ic-egfpt表达的crrna指导内源的crispr/cas系统对phm1-egfp质粒进行切割(见图2b),使得转化后的菌落在含有壮观霉素抗性(100mg/l)的psa平板上不能生长。如图2d所示,pseva-ic-egfpt质粒转化后的菌落数远低于pseva-ic-ncrrna转化后得到的菌落数。
51.同时,检测了提供同源片段作为同源重组模板是否可在pxo99a菌株中实现对质粒的编辑。在质粒phm1-egfp的egfp基因上引入终止密码子,并在突变位点上下游选择各
500bp作为同源序列,同时突变pam位点避免被crispr系统的再次切割。具体步骤如下:以phm1-egfp为模板进行扩增(上游同源臂正向引物:5
’-
gaggagctcgaactccagcaggaccatgtgatc-3’,seq id no:16;上游同源臂反向引物:5
’-
gaccctgaagtagtcgtgaccaccctgacctacg-3’,seq id no:17;下游同源臂正向引物:5
’-
gtggtcacgactacttcagggtcagcttgccgtag-3’,seq id no:18;下游同源臂反向引物:5
’-
gacgcggccgccaaggcgtcattatagaggaagcatg-3’,seq id no:19),扩增体系及反应程序详见实施例1。之后通过融合pcr将扩增的上下游两个同源臂融合成一个片段,引物为上游同源臂的正向引物和下游同源臂的反向引物。扩增体系为50μl:5xq5反应缓冲液10μl;10mm dntps 1μl;10μm正向引物2.5μl;10μm反向引物2.5μl;q5high-fidelity dna polymerase 0.5μl;上游同源片段100ng;下游同源片段100ng;加水至50μl。扩增后纯化融合的同源片段,以saci、noti酶切连接的方式将同源片段构建到质粒pseva-ic-egfpt上,得到质粒pseva-ic-egfptd。最终的同源片段核苷酸序列见seq id no:20,相关质粒图谱见图2a。被内源crispr/cas系统切割的质粒dna可以通过同源片段进行同源重组修复,由于同源片段中内源crispr/cas识别位点的pam被突变,修复后的序列则不会被内源crispr/cas系统识别切割,使得转化后的细菌具有质粒的壮观霉素抗性(见图2c)。如图2d所示,将该质粒转入pxo99
aegfp
菌株中,得到的菌落数远大于电转质粒pseva-ic-egfpt。通过荧光显微镜观察进一步确认,电转质粒pseva-ic-egfptd后,约75%的菌落不能观察到gfp荧光,即有75%的细菌质粒发生了基因编辑。之后,挑选了20个单克隆对其进行了pcr扩增和测序验证,证明质粒上的egfp基因确实发生了突变,意味着通过表达crrna并提供同源片段即可利用pxo99a内源的i-c型crispr/cas系统在质粒上实现高效的基因编辑。
52.实施例3.利用内源的crispr/cas系统对pxo99a基因组进行编辑
53.pxo99a中的crispr/cas系统既然可以很好的对菌体中的质粒进行编辑,证明该系统功能是完整的,接下来将其应用到对基因组进行编辑。由于pxo99a基因组中自身的同源重组效率不足以满足同源重组的发生,需要提高同源重组效率才能在基因组中实现正确的编辑。设计了实验来确定利用pxo99a内源的i-c型crispr/cas系统实现基因编辑需要哪些必要组分。
54.本发明选择突变后会造成菌落呈现白色的xanb2作为靶基因,并设计34bp的间隔序列,载体构建步骤如下:将带有末端接头的引物5
’-
aaaccagcacgctggcctgatacggctgcaacggcacg-3’(seq id no:21)和5
’-
cgaccgtgccgttgcagccgtatcaggccagcgtgctg-3’(seq id no:22)进行梯度退火,后连接到bsai酶切后的质粒pseva-ic-crrna上,得到质粒pseva-ic-xanb2t。在xanb2基因两端分别选取约500bp的片段进行扩增(上游同源臂正向引物:5
’-
cgggatcctctatcgtaactgcacgtaatgccccctc-3’,seq id no:23;上游同源臂反向引物:5
’-
cgctcaatgaaggatgctgtacgtgcggatgctg-3’,seq id no:24;下游同源臂正向引物:5
’-
acagcatccttcattgagcgtctcgttgttgcgtg-3’,seq id no:25;下游同源臂反向引物:5
’-
cgggatccacgaatgcacgcgcatggcag-3’,seq id no:26),扩增体系及反应程序详见实施例1。之后将上下游同源片段通过融合pcr的方法得到上下游同源臂融合的约1000bp片段(相关的核苷酸序列如seq id no:27所示)。扩增后的片段纯化后通过bamhi和hindiii酶切连接后构建到质粒pseva-ic-xanb2t上,最终得到的质粒pseva-ic-xanb2td用于以同源重组的方式实现对xanb2基因的敲除。首先将phm1质粒或phm1-λ-red重组系统转入pxo99a中,制备
电击感受态细胞,之后分别将pseva-ic系列载体转入相应的感受态细胞中,复苏涂板并培养2-3天后,即可通过菌落颜色对突变效率进行统计。如图3b所示,pxo99a内源的i-c型crispr/cas系统在表达正确的crrna、同源片段存在但λ-red重组系统不存在的情况下,仍有少量菌落产生,但全为正常的黄色菌落,无编辑效率。只有在λ-red重组系统同时存在时,pxo99a内源的i-c型crispr/cas系统才可实现高效的基因敲除,效率达到约90%。之后通过鉴定引物xanb2-confirm-f(5
’-
ggtgtcaaagattcagtccgttgag-3’,seq id no:35)和xanb2-confirm-r(5
’-
caagtggttcatctaccgcttcac-3’,seq id no:36)进行pcr鉴定,证明xanb2基因被敲除。测序结果也证明了这一点。
55.实施例4.crrna与dna间的错配对编辑效率的影响
56.在已有的crispr/cas系统相关研究中已经证明,核心序列(seed sequence)对于cas蛋白正确识别原间隔序列至关重要。为了探究i-c型crispr/cas系统对crrna与dna间错配的容忍度,如图4a所示,以pam近端第一位碱基设定为1,对设计的xanb2间隔序列每6个碱基进行突变,分别为m1-6、m7-12、m13-18、m19-24、m25-30及3’端(pam远端)4个碱基突变的m31-34。之后分别将突变后的间隔序列同xanb2的同源片段构建到质粒pseva-ic-crrna上,载体构建同实施例3。并转化至含有phm1-λ-red质粒的pxo99a中,鉴定相应位点突变后对xanb2编辑效率的影响。如图4b所示,仅突变3’端的4个碱基不会改变基因的突变效率,其余5种突变方式的基因编辑都仅是通过λ-red重组系统实现的同源重组编辑。之后继续逐个增加3’端的突变碱基数,随着突变碱基数的增加,基因编辑效率有所下降,当突变到第27位,即3’端第8个碱基后,编辑效率与通过λ-red重组系统实现同源重组编辑的效率一致。3’端4个碱基的错配不会影响pxo99a内源的i-c型crispr/cas基因编辑系统的编辑效率,最多可容忍7个碱基的错配。
57.以上所述的具体实施例,对本发明的目的、技术方案和有益效果进行了进一步详细说明,应理解的是,以上所述仅为本发明的具体实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1