一种吡咯化合物的制备方法与流程
1.本发明涉及一种吡咯化合物的制备方法,属于药物化学技术领域。
背景技术:
2.伏诺拉生,结构式如下式06所示,是一种胃酸分泌抑制剂,具有速效、强劲、持久的胃酸分泌抑制作用,且对胃酸分泌还具有提前终止作用。
3.现有技术中,伏诺拉生的合成路线反应步骤多,产率较低,不利于放大生产。在现有的合成伏诺拉生的路线中,式03所示化合物是合成伏诺拉生的至关重要的中间体。
[0004][0005]
为此本发明提出了一种新的合成路线,可以高效合成伏诺拉生的中间体式03所示化合物。
[0006]
通过化合物03,本发明还提供了一种伏诺拉生的合成路线。本发明提供的伏诺拉生合成路线较短,工艺简便,对环境污染小,工艺安全稳定,利于放大生产。
技术实现要素:
[0007]
发明人经过研究,开发了一种制备化合物03的方法。本发明还提供了一种使用化合物03经过反应,制备化合物06的方法,所述方法步骤较少,操作简便安全。
[0008]
一方面,本发明提供了一种制备化合物03的方法,包括:在有机溶剂中,化合物02在钯催化剂和氢气作用下进行关环反应,反应完毕,经过后处理,制备得到化合物03,
[0009][0010]
关环反应中,所述有机溶剂包括选自1,4-二氧六环,dmf,四氢呋喃中的至少一种。
[0011]
关环反应中,在一些实施方式中,所述有机溶剂包括1,4-二氧六环;在一些实施方式中,所述有机溶剂包括dmf;在一些实施方式中,所述有机溶剂包括四氢呋喃。在一些实施例中,关环反应中,所述有机溶剂为1,4-二氧六环。
[0012]
关环反应中,每一克化合物02,所述有机溶剂的用量可为3ml-10ml。
[0013]
所述关环反应中,反应温度可为40℃-100℃。
[0014]
所述钯催化剂可以为钯碳,氢氧化钯,氯化钯,或其组合。在一些实施方式中,所述钯催化剂为钯碳。
[0015]
所述钯催化剂与化合物02的质量比可为0.05%-10%。在一些实施方式中,所述钯催化剂与化合物02的质量比为0.1%-10%。在一些实施方式中,所述钯催化剂与化合物02的质量比为1%-10%。在一些实施方式中,所述钯催化剂与化合物02的质量比为5%。
[0016]
所述从化合物02制备得到化合物03的后处理包括:反应液降温后过滤,滤液搅拌下与水混合,降温析晶,过滤,干燥,得到化合物03。在一些实施方式中,所述后处理包括:反应液降温至-5℃-40℃,过滤,滤液搅拌下与水混合、析晶,再降温至-5℃-15℃后过滤,固体干燥,得到化合物03。在一些实施方式中,所述后处理包括:反应液降温至0℃-40℃后抽滤,滤液搅拌下加水析晶,再降温至-5℃-5℃后抽滤,固体干燥,得到化合物03。在一些实施方式中,所述后处理包括:反应液降温至20℃-40℃后抽滤,滤液搅拌下加水析晶,再降温至-5℃-5℃后抽滤,固体干燥,得到化合物03。
[0017]
在一些实施方式中,一种制备化合物03的方法,包括:在有机溶剂中,化合物02在钯催化剂和氢气作用下,在40℃-100℃进行关环反应,反应完毕,将反应液降温至0℃-40℃,过滤,滤液搅拌下加水析晶,再降温至-5℃-15℃后过滤,固体干燥,经过后处理,制备得到化合物03。
[0018]
在一些实施方式中,一种制备化合物03的方法,包括:在1,4-二氧六环中,化合物02在钯碳和氢气作用下,在40℃-100℃进行关环反应,反应完毕,经过后处理,制备得到化合物03;所述后处理包括:将反应液降温至0℃-40℃,过滤,滤液搅拌下加入水析晶,再降温至-5℃-5℃后过滤,固体干燥,得到化合物03。
[0019]
所述化合物02的制备方法,包括:在有机反应溶剂中,在相转移催化剂和任选碱试剂条件下,化合物01与丙睛进行反应,反应完毕,经过后处理,制备得到化合物02,
[0020][0021]
所述碱试剂包括选自碳酸钠,碳酸钾,n,n-二异丙基乙胺中的至少一种。在一些实施方式中,所述碱试剂包括碳酸钠;在一些实施方式中,所述碱试剂包括碳酸钾;在一些实施方式中,所述碱试剂包括n,n-二异丙基乙胺。在一些实施例中,所述碱试剂为碳酸钠,有利于反应进行和处理。
[0022]
所述碱试剂与化合物01的摩尔比可以为1:1-2.5:1。在一些实施方式中,所述碱试剂与化合物01的摩尔比为1:1-2:1。在一些实施方式中,所述碱试剂与化合物01的摩尔比为1.1:1-1.5:1。
[0023]
所述相转移催化剂包括四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵、三辛基甲基氯化铵、苄基三乙基氯化铵,或其组合。在一些实施例中,所述催化剂为四丁基溴化铵,有利于反应进行和目标产物的获得。所述相转移催化剂与化合物01的投料摩尔比可以为0.01:1-0.5:1。在一些实施方式中,所述相转移催化剂与化合物01的投料摩尔比为0.05:1-0.2:1。
[0024]
所述制备化合物02所采用的有机反应溶剂,包括选自乙酸乙酯,乙腈,四氢呋喃中的至少一种。在一些实施方式中,所述有机反应溶剂包括乙酸乙酯;在一些实施方式中,所述有机反应溶剂包括乙腈;在一些实施方式中,所述有机反应溶剂包括四氢呋喃。在一些实施例中,所述有机反应溶剂为乙酸乙酯,有利于操作和目标产物的获得。
[0025]
每一克化合物01,所述有机反应溶剂的用量可为3ml-15ml或者5ml-10ml。
[0026]
化合物01与丙睛进行反应,可在20℃-60℃条件下进行。在一些实施方式中,化合物01与丙睛进行反应,在30℃-50℃条件下进行。
[0027]
从化合物01制备得到化合物02的过程中经过后处理,所述后处理包括:降温后加入水,萃取得到有机相并浓缩,加入甲苯、二甲苯或其组合溶剂,升温,打浆,然后降温,过滤,干燥,得到化合物02。
[0028]
在一些实施方式中,所述后处理包括:反应液降温至-5℃-40℃后加入水,萃取分液得到有机相,有机相浓缩,加入甲苯、二甲苯或其组合溶剂,升温至40℃-100℃,打浆0.5h-4h,然后降温至-5℃-25℃,过滤,固体干燥,得到化合物02。在一些实施方式中,所述后处理包括:降温至20℃-40℃后加入水,萃取分液得到有机相,再减压浓缩,加入甲苯、二甲苯或其组合溶剂,升温至70℃-90℃,打浆1h-3h,然后降温至-5℃-15℃,过滤,固体干燥,得到化合物02。在一些实施方式中,降温至0℃-10℃,过滤,固体干燥,得到化合物02。
[0029]
在一些实施方式中,一种化合物02的制备方法包括:在有机反应溶剂中,在相转移催化剂和碱试剂条件下,化合物01与丙睛在0℃-60℃进行反应,反应完毕,反应液降温至0℃-40℃后加入水,萃取分液得到有机相,有机相浓缩,加入甲苯或二甲苯或其组合溶剂,升温至40℃-100℃打浆0.5h-4h,然后降温至-5℃-25℃,过滤,固体干燥,制备得到化合物02。
[0030]
在一些实施方式中,一种化合物02的制备方法包括:在乙酸乙酯中,在四丁基溴化铵和碳酸钠条件下,化合物01与丙睛在20℃-50℃进行反应,反应完毕,经过后处理,制备得到化合物02;所述后处理包括:反应液降温至0℃-40℃后加入水,萃取分液得到有机相,有机相减压浓缩至干,加入甲苯或二甲苯或其组合溶剂,升温至40℃-100℃,打浆0.5h-4h,然后降温至-5℃-15℃,过滤,固体干燥,得到化合物02。
[0031]
在一些实施方式中,一种化合物03的制备方法,包括:在乙酸乙酯中,在四丁基溴化铵和碳酸钠条件下,化合物01与丙睛在20℃-50℃进行反应,反应完毕,经过后处理,制备得到化合物02;所述后处理包括:反应液降温至0℃-40℃后加入水,萃取分液得到有机相,有机相浓缩,加入甲苯或二甲苯或其组合,升温至40℃-100℃,打浆0.5h-4h,然后降温至-5℃-15℃,过滤,固体干燥,得到化合物02;在有机溶剂中,化合物02在钯碳和氢气作用下,在40℃-100℃进行关环反应,反应完毕,经过后处理,制备得到化合物03;所述后处理包括:将反应液降温至20℃-40℃,过滤,滤液搅拌下加水析晶,再降温至-5℃-5℃后过滤,固体干燥,得到化合物03。
[0032]
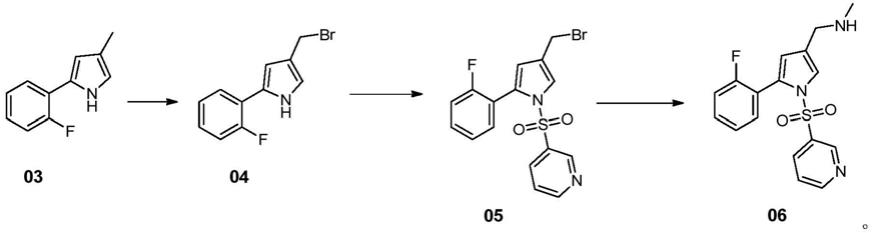
另一方面,本发明提供了一种制备化合物06的方法。一种制备化合物06的方法,包括:前述的化合物03经过取代反应,制得化合物04,化合物04经过磺酰化反应,制备得到化合物05,化合物05经过缩合反应,制备得到化合物06,
[0033][0034]
在一些实施方式中,一种制备化合物06的方法,包括:按照前述的制备化合物03的方法制备得到化合物03,化合物03经过取代反应,制得化合物04,化合物04经过磺酰化反应,制备得到化合物05,化合物05经过缩合反应,制备得到化合物06。
[0035]
所述化合物04的制备方法可包括:在有机溶剂中,在偶氮二异丁腈作用下,前述的化合物03通过与n-溴代琥珀酰亚胺经过取代反应,经过后处理,制备得到化合物04。
[0036]
取代反应中,所述有机溶剂包括选自乙腈,二氯乙烷中的至少一种。在一些实施方式中,所述有机溶剂包括乙腈;在一些实施方式中,所述有机溶剂包括二氯乙烷。在一些实施例中,所述有机溶剂为乙腈。
[0037]
取代反应中,每一克化合物03,所述有机溶剂用量可为3ml-20ml,或者5ml-15ml。
[0038]
所述偶氮二异丁腈与化合物03的投料摩尔比可为0.9:1-1.1:1。
[0039]
所述n-溴代琥珀酰亚胺与化合物03的投料摩尔比可以为1:1-2.5:1。在一些实施方式中,所述n-溴代琥珀酰亚胺与化合物03的投料摩尔比为1.2:1-2:1。
[0040]
所述取代反应的反应温度可以控制为20℃-80℃或者40℃-80℃或者20℃-40℃。
[0041]
取代反应反应完毕后,所述后处理包括:反应液中加入水和二氯甲烷,萃取分液得到有机相,有机相浓缩,所得物与乙腈、乙酸乙酯、丙酮、甲醇或其组合溶剂混合,打浆0.5小时-4小时,控温-5℃-30℃,过滤,固体干燥,得到化合物04。在一些实施方式中,所述后处理包括:反应液降温至40℃以下,加入水和二氯甲烷,萃取分液得到有机相,有机相减压浓缩,所得物与乙腈、乙酸乙酯、丙酮、甲醇或其组合溶剂混合,在40℃-80℃打浆0.5小时-4小时,降温至-5℃-20℃,过滤,固体干燥,得到化合物04。
[0042]
在一些实施方式中,一种化合物04的制备方法,包括:在有机溶剂中,在偶氮二异丁腈作用下,化合物03与n-溴代琥珀酰亚胺在0℃-80℃进行取代反应,反应完毕后,反应液控温为0℃-40℃,加入水和萃取溶剂,萃取分液得到有机相并浓缩,所得物与乙腈、乙酸乙酯、丙酮、甲醇或其组合混合,打浆0.5小时-4小时,控温-5℃-20℃,过滤,固体干燥,得到化合物04。
[0043]
在一些实施方式中,一种化合物04的制备方法,包括:在乙腈中,在偶氮二异丁腈作用下,前述的化合物03通过n-溴代琥珀酰亚胺在0℃-80℃进行取代反应,反应完毕后,反应液控温为0-40℃,加入水和二氯甲烷,萃取分液得到有机相,减压浓缩,所得物与乙腈混合,打浆0.5小时-4小时,控温-5℃-15℃,过滤,固体干燥,得到化合物04。
[0044]
所述化合物05的制备方法包括:在有机溶剂中,在有机碱和4-二甲氨基吡啶条件下,化合物04与吡啶磺酰氯经过磺酰化反应,制备得到化合物05。
[0045]
磺酰化反应中,所述有机溶剂包括选自二氯甲烷,四氢呋喃中的至少一种。在一些实施方式中,所述有机溶剂包括二氯甲烷;在一些实施方式中,所述有机溶剂包括四氢呋
喃。在一些实施例中,所述有机溶剂为二氯甲烷。
[0046]
每一克化合物04,所述有机溶剂的用量可为3ml-20ml或者5ml-15ml。
[0047]
化合物04与吡啶磺酰氯的投料摩尔比可以为1:1-1:1.5,或者1:1-1:1.3。
[0048]
所述有机碱为三乙胺,n,n-二异丙基乙基胺,吡啶,异丙基胺或其组合。所述有机碱与化合物04的投料摩尔比可为1:1-3:1或1.2:1-2:1。
[0049]
所述磺酰化反应的反应温度可为0℃-60℃,或者20℃-40℃,或者20℃-60℃,或者30℃-60℃。
[0050]
磺酰化反应反应完毕后,可经过后处理,得到化合物05;所述后处理包括:反应液中加入水,分离得到有机相并浓缩,所得物与乙腈、乙酸乙酯、丙酮、甲醇或其组合溶剂混合,打浆0.1-2小时,降温至-5℃-15℃,过滤,固体干燥,得到化合物05。在一些实施方式中,磺酰化反应反应完毕后,经过后处理,所述后处理包括:反应液中加入水,分离得到有机相并浓缩,所得物与乙腈、乙酸乙酯、丙酮、甲醇或其组合溶剂混合,在40℃-80℃打浆0.1-2小时,降温至-5℃-5℃,过滤,固体干燥,得到化合物05。
[0051]
在一些实施方式中,一种化合物05的制备方法,包括:在有机溶剂中,在有机碱和4-二甲氨基吡啶条件下,化合物04与吡啶磺酰氯在0℃-60℃经过磺酰化反应,反应完毕;反应液中加入水,分离得到有机相并浓缩,所得物与乙腈、乙酸乙酯、丙酮、甲醇或其组合溶剂混合,打浆0.1小时-2小时,降温至-5℃-5℃,过滤,固体干燥,得到化合物05。
[0052]
在一些实施方式中,一种化合物05的制备方法,包括:在有机溶剂中,在三乙胺和4-二甲氨基吡啶条件下,化合物04与吡啶磺酰氯在0℃-60℃经过磺酰化反应,反应完毕,经过后处理,制备得到化合物05;所述后处理包括:反应液中加入水,分离得到有机相并浓缩,所得物与乙腈混合,打浆0.1-2小时,降温至-5℃-5℃,过滤,固体干燥,得到化合物05。
[0053]
所述化合物06的制备方法包括:在有机溶剂中,在碳酸钾和碘化钾作用下,化合物05与甲胺或其盐酸盐经过缩合反应,制备得到化合物06。
[0054]
缩合反应中,所述有机溶剂包括选自二氯甲烷,dmf,dmac,乙腈,四氢呋喃,乙酸乙酯中的至少一种。在一些实施方式中,所述有机溶剂包括二氯甲烷;在一些实施方式中,所述有机溶剂包括dmf;在一些实施方式中,所述有机溶剂包括dmac;在一些实施方式中,所述有机溶剂包括乙腈;在一些实施方式中,所述有机溶剂包括四氢呋喃;在一些实施方式中,所述有机溶剂包括乙酸乙酯。在一些实施例中,所述有机溶剂为二氯甲烷。
[0055]
缩合反应中,每一克化合物05,所述有机溶剂的用量可以为5ml-25ml,或者10ml-20ml,或者8ml-15ml。
[0056]
化合物05与甲胺或其盐酸盐的投料摩尔比可以为1:1-1:2.5或者1:1-1:2。
[0057]
所述化合物05与碳酸钾的投料摩尔比可以为1:1-1:2.5。在一些实施方式中,所述化合物05与碳酸钾的投料摩尔比可以为1:1-1:2。
[0058]
所述碘化钾与化合物05的投料摩尔比可以为1:1-2:1。在一些实施方式中,所述碘化钾与化合物05的投料摩尔比为1:1.1-1.6:1。
[0059]
所述缩合反应的反应温度可以为30℃-100℃。在一些实施方式中,所述缩合反应的反应温度为40℃-60℃,或者50℃-80℃,或者60℃-100℃。
[0060]
缩合反应反应完毕后,可以经过后处理,得到化合物06,所述后处理包括:反应液中加入水,萃取分液,水相再用萃取溶剂萃取分液,合并有机相并浓缩,加入正己烷、环己
烷、异丙醇或其组合溶剂打浆,降温至-5℃-30℃,过滤,固体干燥,得到化合物06。在一些实施方式中,所述后处理包括:反应液控温0℃-30℃,加入水,萃取分液,水相再用萃取溶剂萃取分液,合并有机相并浓缩,加入正己烷、环己烷、异丙醇或其组合溶剂,在30℃-70℃打浆0.5小时-4小时,降温至-5℃-20℃,过滤,固体干燥,得到化合物06。在一些实施方式中,所述后处理包括:反应液控温0℃-30℃加入水,萃取分液,水相再用二氯甲烷萃取分液,合并有机相并浓缩,加入正己烷在30℃-70℃打浆0.5小时-4小时,降温至-5℃-10℃,过滤,固体干燥,得到化合物06。
[0061]
在一些实施方式中,一种化合物06的制备方法包括:在有机溶剂中,在碳酸钾和碘化钾作用下,化合物05与甲胺或其盐酸盐在30℃-100℃经过缩合反应,反应完毕;反应液中加入水,萃取分液,水相再用萃取溶剂萃取分液,合并有机相并浓缩,加入正己烷、环己烷、异丙醇或其组合溶剂,在30℃-70℃打浆0.5小时-4小时,降温至-5℃-30℃,过滤,固体干燥,得到化合物06。
[0062]
在一些实施方式中,一种化合物06的制备方法包括:在有机溶剂中,在碳酸钾和碘化钾作用下,化合物05与甲胺盐酸盐在30℃-100℃经过缩合反应,反应完毕,经过后处理,制备得到化合物06;所述后处理包括:反应液控温-5℃-30℃加入水,萃取分液,水相再用萃取溶剂萃取分液,合并有机相并浓缩,加入正己烷打浆,降温至-5℃-10℃,过滤,固体干燥,得到化合物06。
[0063]
在一些实施方式中,一种化合物06的制备方法包括:
[0064]
(1)在有机反应溶剂中,在相转移催化剂和碱试剂条件下,化合物01与丙睛在0℃-60℃进行反应,反应完毕;反应液降温至-5℃-40℃后加入水,萃取分液得到有机相,有机相浓缩,加入甲苯或二甲苯或其组合溶剂,升温至40℃-100℃,打浆0.5h-4h,然后降温至-5℃-25℃,过滤,固体干燥,制备得到化合物02;
[0065]
(2)在有机溶剂中,化合物02在钯催化剂和氢气作用下,在40℃-100℃进行关环反应,反应完毕;将反应液降温至-5℃-40℃,过滤,滤液搅拌下加水析晶,再降温至-5℃-15℃后过滤,固体干燥,经过后处理,制备得到化合物03;
[0066]
(3)在有机溶剂中,在偶氮二异丁腈作用下,化合物03与n-溴代琥珀酰亚胺在0℃-80℃进行取代反应,反应完毕;反应液控温为-5℃-40℃,加入水和萃取溶剂,萃取分液得到有机相并浓缩,所得物与乙腈、乙酸乙酯、丙酮或其组合溶剂混合,打浆0.5小时-4小时,控温-5℃-30℃,过滤,固体干燥,得到化合物04;
[0067]
(4)在有机溶剂中,在有机碱和4-二甲氨基吡啶条件下,化合物04与吡啶磺酰氯在0℃-60℃经过磺酰化反应,反应完毕;反应液中加入水,分液得到有机相并浓缩,所得物与乙腈、乙酸乙酯、丙酮或其组合溶剂混合,打浆0.1小时-2小时,降温至-5℃-15℃,过滤,固体干燥,得到化合物05;
[0068]
(5)在有机溶剂中,在碳酸钾和碘化钾作用下,化合物05与甲胺或其盐酸盐在30℃-100℃经过缩合反应,反应完毕;反应液控温为-5℃-30℃加入水,萃取分液,水相再用萃取溶剂萃取分液,合并有机相并浓缩,加入正己烷、环己烷、异丙醇或其组合溶剂,在30℃-70℃打浆0.5小时-4小时,降温至-5℃-30℃,过滤,固体干燥,得到化合物06。
[0069]
在一些实施方式中,一种化合物06的制备方法包括:
[0070]
(1)在乙酸乙酯中,在四丁基溴化铵和碳酸钠条件下,化合物01与丙睛在20℃-50
℃进行反应,反应完毕,反应液降温至0℃-40℃后加入水,萃取分液得到有机相,有机相浓缩,加入甲苯,升温至40℃-100℃,打浆0.5h-4h,然后降温至-5℃-25℃,过滤,固体干燥,制备得到化合物02;
[0071]
(2)在1,4-二氧六环中,化合物02在钯碳和氢气作用下,在40℃-100℃进行关环反应,反应完毕,将反应液降温至0℃-40℃,过滤,滤液搅拌下加水析晶,再降温至-5℃-15℃后过滤,固体干燥,经过后处理,制备得到化合物03;
[0072]
(3)在乙腈中,在偶氮二异丁腈作用下,化合物03与n-溴代琥珀酰亚胺在0℃-80℃进行取代反应,反应完毕后,反应液控温为0℃-40℃,加入水和二氯甲烷,萃取分液得到有机相并浓缩,所得物与乙腈混合,打浆0.5小时-4小时,控温-5℃-20℃,过滤,固体干燥,得到化合物04;
[0073]
(4)在二氯甲烷中,在三乙胺和4-二甲氨基吡啶条件下,化合物04与吡啶磺酰氯在0℃-60℃经过磺酰化反应,反应完毕;反应液中加入水,分离得到有机相并浓缩,所得物与乙腈混合,打浆0.1小时-2小时,降温至-5℃-5℃,过滤,固体干燥,得到化合物05;
[0074]
(5)在二氯甲烷中,在碳酸钾和碘化钾作用下,化合物05与甲胺盐酸盐在30℃-100℃经过缩合反应,反应完毕;反应液控温0℃-30℃加入水,萃取分液,水相再用二氯甲烷萃取分液,合并有机相并浓缩,加入正己烷在30℃-70℃打浆0.5小时-4小时,降温至-5℃-10℃,过滤,固体干燥,得到化合物06。
[0075]
上述方法中,涉及萃取时,可以使用的萃取溶剂可为二氯甲烷,甲苯,乙酸乙酯等可以与水分层的有机溶剂。
[0076]
上述方法中,涉及干燥时,将固体干燥至恒重或者干燥前后重量变化不超过
±
1%;涉及浓缩时,可在减压条件下或其他条件下,蒸馏至无明显馏分蒸出。
[0077]
本发明提供的化合物06的制备方法相对简单,工艺路线较短,易于操作实施,适合放大生产。
具体实施方式
[0078]
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例对本发明作进一步的详细说明。
[0079]
本发明所使用的试剂均可以从市场上购得或者可以通过本发明所描述的方法制备而得。
[0080]
本发明中,反应完毕是指原料的剩余量不高于投料量的5%或不高于3%或不高于1%或不高于0.5%;可通过hplc(高效液相色谱法)或tlc(薄层色谱法)进行判断确认。
[0081]
本发明中,室温指环境温度,在15℃-35℃或20℃-30℃或23℃-28℃或25℃。
[0082]
本发明中,g:克;ml:毫升;℃:摄氏度;h:小时;min:分钟;ms表示质谱;hplc表示高效液相色谱。dmf表示n,n-二甲基甲酰胺;dmac表示n,n-二甲基乙酰胺;taba表示四丁基溴化铵;nbs表示n-溴代琥珀酰亚胺;tea表示三乙胺;dmap表示4-二甲氨基吡啶。
[0083]
实施例1
[0084]
化合物02的制备
[0085][0086]
室温下反应瓶中加入100.00g化合物01,33.00g丙腈,121.89g碳酸钠,16.12g四丁基溴化铵,600ml乙酸乙酯,加完升温至40℃,搅拌条件下反应4h,取样检测,hplc结果显示原料反应完全,停止反应;降温至30℃加入250ml水,萃取分液得到有机相,减压至35℃浓缩至干,加入100ml甲苯升温至80℃打浆2h,降温至20℃抽滤,滤饼在80℃条件下真空干燥12h,得到化合物01黄色固体产物76.11g,收率86.40%,纯度96.31%。
[0087]
ms:[m+1]=192.11;
[0088]
核磁1h nmr(400mhz,dmso)δ7.89-7.96(m,2h),7.25-7.36(m,2h),3.21(m,j=14.3hz,1h),2.66-3.03(dd,2h),1.51(t,3h)。
[0089]
实施例2
[0090]
化合物03的制备
[0091][0092]
室温下反应瓶中加入70.00g化合物02,10%钯碳3.50g,1,4-二氧六环250ml,加完置换氢气,升温至80℃,搅拌条件下反应16h,取样检测,hplc结果显示原料反应完全,停止反应;降温至30℃抽滤,滤液搅拌下加入250ml水析晶,加完降温至10℃抽滤,滤饼80℃条件下真空干燥12h,得到化合物03黄色固体产物46.30g,收率72.18%,纯度97.22%。
[0093]
ms:[m+1]=176.11;
[0094]
核磁1h nmr(400mhz,dmso)δ9.79(s,1h),7.69-7.81(m,2h),7.52(m,1h),7.33(m,1h),6.71(s,1h),6.35(s,1h),2.12(s,3h)。
[0095]
实施例3
[0096]
化合物04的制备
[0097][0098]
室温下反应瓶中加入50.00g化合物03,103.23g nbs,偶氮二异丁腈47.62g,500ml乙腈,加完升温至80℃,搅拌条件下反应16h,取样检测,hplc结果显示原料反应完全,停止反应;降温至30℃加入500ml水和500ml二氯甲烷,萃取分液得到有机相,减压至35℃浓缩至干,加入200ml乙腈升温至60℃打浆2h,降温至20℃抽滤,滤饼60℃条件下真空干燥12h,得到化合物04黄色固体产物45.55g,收率62.81%,纯度95.44%。
[0099]
ms:[m+1]=254.05;
[0100]
核磁1h nmr(400mhz,dmso)δ10.20(s,1h),7.69-7.81(m,2h),7.52(m,1h),7.33(m,1h),6.71(s,1h),6.35(s,1h),4.52(s,2h)。
[0101]
实施例4
[0102]
化合物05的制备
[0103][0104]
室温下反应瓶中加入50.00g化合物04,吡啶-3-磺酰氯46.18g,三乙胺40.48g,4-二甲氨基吡啶2.44g,500ml二氯甲烷,加完升温至40℃,搅拌条件下反应8h,取样检测,hplc结果显示原料反应完全,停止反应;降温至30℃加入500ml水,萃取分液得到有机相,减压至35℃浓缩至干,加入200ml乙腈升温至50℃打浆1h,降温至0℃抽滤,滤饼在60℃条件下真空干燥12h,得到化合物05黄色固体产物64.32g,收率83.36%,纯度96.43%。
[0105]
ms:[m+1]=395.00;
[0106]
核磁1h nmr(400mhz,dmso)δ8.89(d,1h),8.64(s,1h),7.85(dd,1h),7.76(s,1h),7.55-7.64(m,1h),7.49-7.54(m,1h),7.29-7.38(m,2h),7.17-7.25(m,2h),7.00-7.05(m,1h),6.41-6.46(d,1h),4.57-4.65(s,2h)。
[0107]
实施例5
[0108]
化合物06的制备
[0109][0110]
室温下反应瓶中加入50.00g化合物05,甲胺盐酸盐17.56g,碳酸钾35.93g,碘化钾33.20g,600ml二氯甲烷,加完升温至40℃,搅拌条件下反应8h,取样检测,hplc结果显示原料反应完全,停止反应;降温至30℃加入500ml水,萃取分液,水相再加500ml二氯甲烷萃取分液,合并有机相,减压至35℃浓缩至干,加入250ml正己烷升温至60℃打浆2h,降温至20℃抽滤,滤饼80℃条件下真空干燥12h,得到化合物06黄色固体产物35.96g,收率82.31%,纯度98.34%。
[0111]
ms:[m+1]=346.15;
[0112]
核磁1h nmr(400mhz,dmso)δ8.79(d,1h),8.62(s,1h),7.89(dd,1h),7.78(s,1h),7.60-7.64(m,1h),7.49-7.54(m,1h),7.28-7.38(m,2h),7.15-7.25(m,2h),7.02-7.07(m,1h),6.40-6.46(d,1h),3.77-3.85(s,2h),2.51-3.60(s,3h)。
[0113]
本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在本发明内容、
精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1