一种盐酸左旋咪唑的合成方法与流程
1.本发明涉及有机合成领域,尤其涉及一种盐酸左旋咪唑的合成方法。
背景技术:
2.盐酸左旋咪唑(levamisole hydrochloride),cas:16595-80-5,化学名:(s)-6-苯基-2,3,5,6-四氢-咪唑[2,1-b]噻唑单盐酸盐,可用于治疗蛔虫、钩虫、蛲虫和粪类圆线虫感染。盐酸左旋咪唑是盐酸四咪唑的左旋体,其活性为盐酸四咪唑(消旋体)的1-2倍,且毒副作用更低。此外盐酸左旋咪唑还有免疫调节和免疫兴奋功能。
[0003]
中国医药工业杂志,1994,25(8):342.报道盐酸左旋咪唑的制备方法为:以盐酸四咪唑为原料,中和得到四咪唑,通过使用手性酸拆分剂(如n-对甲苯磺酰-l-(+)-谷氨酸单钠盐)将左旋咪唑(四咪唑的左旋体)分离出来,然后与盐酸成盐得到盐酸左旋咪唑。该方法同时得到副产品右旋咪唑,右旋咪唑经过强碱条件消旋为四咪唑后,重新进入拆分工序制备盐酸左旋咪唑。该工艺用到价格较高的手性酸拆分剂,工序冗长,收率较低。
[0004]
山东医药工业,1996,15(2):1-35.报道了消旋体盐酸四咪唑的合成方法,具体路线如下:
[0005][0006]
该路线以氧化苯乙烯为起始物料,经六步反应,以55%总收率制备盐酸四咪唑。起始物料氧化苯乙烯有强烈的刺激性气味,使用该路线会有环境保护和劳动保护方面的问题。
[0007]
总之,使用氧化苯乙烯合成盐酸四咪唑,盐酸四咪唑拆分制备盐酸左旋咪唑的方法存在不利于环保和劳动保护、工序冗长、三废产生量大、产品成本较高的缺点。
[0008]
chem.pharm.bull.43(5):738-747,1995.报道了通过不对称催化氢化直接合成盐酸左旋咪唑的方法,具体路线如下:
[0009][0010]
该方法用到的贵金属铑催化剂[rh(cod)cl]2,所以该方法尚无产业化应用价值。
技术实现要素:
[0011]
本发明针对现有盐酸左旋咪唑制备方法步骤繁琐且收率低的问题,提供一种盐酸左旋咪唑的合成方法,该方法以食品防腐剂l-扁桃酸(cas:17199-29-0)为起始物料,经过酯化、胺解、还原、氯代、缩合、环和、成盐等步骤最终制备出盐酸左旋咪唑,可选择相关联的三个合成路线。
[0012]
本发明合成路线(一)如下:
[0013][0014]
合成步骤为:
[0015]
步骤一、在醇溶液中,将l-扁桃酸(ⅱ)与醇发生酯化反应得化合物(ⅲ);
[0016]
步骤二、化合物(ⅲ)与乙醇胺反应,胺解得化合物(ⅳ);
[0017]
步骤三、在有机溶剂中,化合物(ⅳ)被还原剂还原成化合物(
ⅴ
);
[0018]
步骤四、化合物(
ⅴ
)被氯代试剂氯代得化合物(ⅵ);
[0019]
步骤五、碱性条件下,化合物(ⅵ)在缩合剂的作用下缩合成化合物(
ⅷ
);
[0020]
步骤六、在有机溶剂中,化合物(
ⅷ
)在盐酸中成盐,制得盐酸左旋咪唑。
[0021]
合成路线(二)如下:
[0022][0023]
合成步骤为:
[0024]
步骤一、在醇溶液中,将l-扁桃酸(ⅱ)与醇发生酯化反应得化合物(ⅲ);
[0025]
步骤二、化合物(ⅲ)与乙醇胺反应,胺解得化合物(ⅳ);
[0026]
步骤三、在有机溶剂中,化合物(ⅳ)被还原剂还原成化合物(
ⅴ
);
[0027]
步骤四、化合物(
ⅴ
)被氯代试剂氯代得化合物(ⅵ);
[0028]
步骤五、化合物(ⅵ)在缩合剂的作用下缩合成化合物(ⅶ);
[0029]
步骤六、在碱性溶液中,化合物(ⅶ)环化成化合物(
ⅷ
);
[0030]
步骤七、在有机溶剂中,化合物(
ⅷ
)在盐酸中成盐,制得盐酸左旋咪唑。
[0031]
合成路线(三)如下:
[0032][0033]
合成步骤为:
[0034]
步骤一、在醇溶液中,将l-扁桃酸(ⅱ)与醇发生酯化反应得化合物(ⅲ);
[0035]
步骤二、化合物(ⅲ)与乙醇胺反应,胺解得化合物(ⅳ);
[0036]
步骤三、在有机溶剂中,化合物(ⅳ)被还原剂还原成化合物(
ⅴ
);
[0037]
步骤四、化合物(
ⅴ
)被氯代试剂氯代得化合物(ⅵ);
[0038]
步骤五、在水中,化合物(ⅵ)水解转化为化合物(
ⅸ
);
[0039]
步骤六、化合物(
ⅸ
)在缩合剂的作用下缩合成化合物(
ⅹ
);
[0040]
步骤七、化合物(
ⅹ
)在氯代试剂存在下环化成化合物(
ⅷ
);
[0041]
步骤八、在有机溶剂中,化合物(
ⅷ
)在盐酸中成盐,制得盐酸左旋咪唑。
[0042]
在上述三个合成路线中,r为c
1-c6的烷基,优选甲基、乙基、异丙基、乙烯基或环己基,更优选甲基或乙基;步骤三所用有机溶剂选自四氢呋喃、甲醇、乙醇或异丙醇,优选四氢呋喃;还原剂选自硼烷,能够产生硼烷的试剂组合硼氢化钠/碘、硼氢化钠/三甲基氯硅烷、硼氢化钠/硫酸、硼氢化钠/氯化锌、硼氢化锂或氢化锂铝,优选硼烷;步骤四中所用氯代试剂选自二氯亚砜、三氯氧磷或三氯化磷,优选二氯亚砜。
[0043]
合成路线(一)的步骤五、合成路线(二)的步骤五以及合成路线(三)的步骤六所用缩合剂选自硫脲或硫氰化钾,优选硫脲。
[0044]
合成路线(一)的步骤六、合成路线(二)的步骤七以及合成路线(三)的步骤八所用有机溶剂选自乙腈、甲醇、乙醇、水、丙酮或四氢呋喃,优选乙腈。
[0045]
合成路线(一)的步骤五反应条件为碱性溶液,碱性溶液为氢氧化钠溶液、氢氧化钾溶液、碳酸钠或碳酸钾溶液;合成路线(二)的步骤六所用碱性溶液为氢氧化钠溶液、氢氧化钾溶液、碳酸钠或碳酸钾溶液。
[0046]
合成路线(三)步骤五反应条件为水溶液,该步骤中化合物ⅵ在水中水解即可得到化合物
ⅸ
,加入碱如氢氧化钠、氢氧化钾,起到中和化合物
ⅸ
盐酸盐的作用,以便使游离的化合物
ⅸ
进入有机相。
[0047]
合成路线(三)的步骤七所用氯代试剂选自二氯亚砜、三氯氧磷或三氯化磷,优选二氯亚砜。
[0048]
本发明的有益效果是:与氧化苯乙烯合成盐酸四咪唑、盐酸四咪唑拆分制备盐酸左旋咪唑的方法相比,本发明合成方法避免使用刺激性气味的氧化苯乙烯,具有环保和劳动保护方面的优势;不经过拆分步骤,直接合成盐酸左旋咪唑,克服了拆分步骤工序冗长、三废产生量大、产品成本较高的缺点。与不对称催化氢化合成盐酸左旋咪唑方法相比,避免了昂贵手性催化剂的使用,成本大幅度降低,不需要使用催化氢化反应,提高了工艺安全性。本发明合成方法采用廉价易得的手性l-扁桃酸为起始物料,整条路线原材料成本低、绿色环保,克服了目前盐酸左旋咪唑拆分方法、不对称合成方法的缺点。
附图说明
[0049]
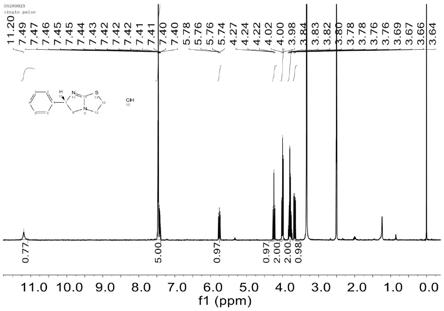
图1为本发明所得盐酸左旋咪唑的氢谱;
[0050]
图2为本发明所得盐酸左旋咪唑的质谱。
具体实施方式
[0051]
以下结合实例对本发明进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
[0052]
实施例1
[0053]
采用合成路线(一)制备盐酸左旋咪唑,具体步骤为:
[0054]
1、化合物(ⅲ)的合成
[0055]
1l反应瓶中,加入100.0g l-扁桃酸(ⅱ),300.0g甲醇,1.1g硫酸,升温至65℃,回流反应2h;减压脱除溶剂,加入150g甲苯,碳酸氢钠溶液(1.8g碳酸氢钠溶解于50g水中),分液,甲苯相用50g水洗一次;减压脱除甲苯,得到无色液体,冷却至室温变为白色固体,l-扁桃酸甲酯,108.1g,收率99%;
[0056]
2、化合物(ⅳ)的合成
[0057]
500ml反应瓶中,加入100.0g l-扁桃酸甲酯,81.9g乙醇胺,升温至90℃,反应2h,减压脱除溶剂,得到无色油状物,冷却至室温变为白色固体,115.1g,收率98%;
[0058]
3、化合物(
ⅴ
)的合成
[0059]
2l反应瓶中,加入50.0g化合物(ⅳ),冰水浴下,滴加538ml硼烷四氢呋喃溶液(1mol/l),滴加完毕,升温至65℃反应12h。冷却至室温,滴加100g甲醇,150g盐酸,升温至65℃,反应1h,减压脱除溶剂,得到白色固液混合物,加入100g乙酸乙酯打浆,过滤,得到白色固体化合物(
ⅴ
)47.4g,收率85%;
[0060]
4、化合物(ⅵ)的合成
[0061]
500ml反应瓶中,加入40.0g化合物(
ⅴ
),175.0g氯化亚砜,升温至60-70℃,反应3h,减压脱除溶剂,得到淡黄色油状物,加入40g乙腈,60g乙酸乙酯,降温至-10℃析晶,得到近白色固体化合物(ⅵ)41.6g,收率89%;
[0062]
5、化合物(
ⅷ
)的合成
[0063]
50ml反应瓶中,加入2.0g化合物(ⅵ),30g乙醇,0.7g硫脲,1.0g氢氧化钠,升温至80-90℃反应5h,减压脱除溶剂,得到黄色固液混合物,硅胶柱层析,得到黄色固体化合物(
ⅷ
)1.4g,收率88%;
[0064]
6、盐酸左旋咪唑(ⅰ)的合成
[0065]
100ml反应瓶中,加入12.0g化合物(
ⅷ
),55g乙腈,20-30℃下,滴加6.6g盐酸,搅拌反应2h,降温至0-10℃,过滤,得到白色固体盐酸左旋咪唑13.3g,收率94%。
[0066]
实施例2
[0067]
采用合成路线(二)制备盐酸左旋咪唑,具体步骤为:
[0068]
步骤1-4与实施例1步骤1-4相同;
[0069]
5、化合物(ⅶ)的合成
[0070]
100ml反应瓶中,加入20.0g化合物(ⅵ),6.6g硫脲,80g水,升温至90-100℃,反应2h,浓缩脱水,析出淡黄色固体,过滤,得到淡黄色固体,19.6g,收率90%;
[0071]
或者,
[0072]
100ml反应瓶中,加入20.0g化合物(ⅵ),9.9g硫氰化钾,25g乙醇,75g水,升温至回流,反应12h;浓缩脱溶剂,析出淡黄色固体,过滤,得到淡黄色固体,20.1g,收率92%;
[0073]
6、化合物(
ⅷ
)的合成
[0074]
250ml反应瓶中,加入18.0g化合物(ⅶ),80g水,80g乙醇,3.1g氢氧化钠,升温至回流反应2h,减压脱除乙醇,降温至降温至20-30℃,过滤,得到淡黄色固体12.2g,收率92%;
[0075]
7、盐酸左旋咪唑(ⅰ)的合成
[0076]
100ml反应瓶中,加入12.0g化合物(
ⅷ
),55g乙腈,20-30℃下,滴加6.6g盐酸,搅拌
反应2h,降温至0-10℃,过滤,得到白色固体13.3g,收率94%。
[0077]
实施例3
[0078]
采用合成路线(三)制备盐酸左旋咪唑,具体步骤为:
[0079]
1、化合物(ⅲ)的合成
[0080]
1l反应瓶中,加入50.0g l-扁桃酸(ⅱ),150.0g乙醇,10-20℃下,滴加47.0g二氯亚砜,滴加完毕,10-20℃反应1h,减压脱除溶剂,得到无色油状物,加入100g甲苯,50g水,分液取甲苯相,减压脱除溶剂得到无色油状液体,冷却至室温变为白色固体,得化合物(ⅲ)l-扁桃酸乙酯58.0g,收率98%;
[0081]
2、化合物(ⅳ)的合成
[0082]
250ml反应瓶中,加入50.0g l-扁桃酸乙酯,20.5g乙醇胺,70.0g甲苯,升温至100℃,反应2h,减压脱除溶剂,得到无色油状物53.7g,收率99%;
[0083]
3、化合物(
ⅴ
)的合成
[0084]
2l反应瓶中,加入50.0g化合物(ⅳ),450g乙醇,冰水浴下,分批加入15.6g硼氢化锂,升温至回流反应5h,冷却至室温,滴加105g盐酸,减压脱除溶剂,加入150g乙酸乙酯打浆,过滤,得到白色固体44.0g,收率79%;
[0085]
4、化合物(ⅵ)的合成
[0086]
500ml反应瓶中,加入40.0g化合物(
ⅴ
),175.0g氯化亚砜,升温至40-50℃,反应6h,减压脱除溶剂,得到淡黄色油状物,加入40g乙腈,60g乙酸乙酯,降温至-10℃析晶,得到近白色固体40.7g,收率88%;
[0087]
5、化合物(
ⅸ
)的合成
[0088]
250ml反应瓶中,加入11.0g化合物(ⅵ),120g水,升温至回流反应2h,滴加2mol/l氢氧化钠水溶液调节ph=10,二氯甲烷100g萃取,有机相减压脱除溶剂,得到淡黄色油状物,加入50g乙腈溶解,滴加盐酸乙醇溶液成盐,过滤,得到化合物(
ⅸ
)9.7g,收率95%;
[0089]
6、化合物(
ⅹ
)的合成
[0090]
100ml反应瓶中,加入8.7g化合物(
ⅸ
),55g水,3.3g硫脲,回流反应18h,加入5.4g盐酸,继续反应1h,浓缩脱水,过滤,得到淡黄色固体化合物(
ⅹ
)7.6g,收率80%;
[0091]
或者,
[0092]
100ml反应瓶中,加入5.0g化合物(
ⅸ
),10g水,40g乙醇,2.3g硫氰化钾,升温至回流反应2h,加入3.0g盐酸,继续反应1h,减压脱除溶剂,加入23g丙酮,加热溶解,降温至0-10℃,过滤,得到近白色固体化合物(
ⅹ
)4.3g,收率78%;
[0093]
7、化合物(
ⅷ
)的合成
[0094]
100ml反应瓶中,加入3.7g化合物化合物(
ⅹ
),40g二氯甲烷,1.9g二氯亚砜,20-30℃下,搅拌反应3h,反应体系中加入55g水,5.1g碳酸钾,继续搅拌1h。分液,二氯甲烷相50g水洗一次,减压脱除溶剂,得到黄色固体化合物(
ⅷ
)9.2g,收率93%;
[0095]
8、盐酸左旋咪唑(ⅰ)的合成
[0096]
100ml反应瓶中,加入12.0g化合物(
ⅷ
),55g乙腈,20-30℃下,滴加6.6g盐酸,搅拌反应2h,降温至0-10℃,过滤,得到白色固体13.3g,收率94%。
[0097]
图1为本发明所得盐酸左旋咪唑的氢谱:
[0098]1h-nmr(400mhz,dmso-d6)δ11.21(s,1h),7.49
–
7.33(m,5h),5.76(dd,j=10.4,
8.4hz,1h),4.24(t,j=10.3hz,1h),4.00(dd,j=8.2,7.0hz,2h),3.87
–
3.72(m,2h),3.67(dd,j=10.2,8.3hz,1h).
[0099]
图2为本发明所得盐酸左旋咪唑的质谱:
[0100]
ms(esi)m/z=205.28[m+h]
+
。
[0101]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1