烷基蒽醌组合物及其制备方法与流程
1.本发明涉及一种有机物的组合物及其制备方法,具体地说,涉及一种烷基蒽醌的组合物,以及通过蒽来制备烷基蒽醌组合物的方法。
背景技术:
2.烷基蒽醌是蒽醌法制过氧化氢工艺过程中的载体,主要包括乙基蒽醌、戊基蒽醌及其它们的四氢衍生物,其现有生产工艺为苯酐法,该工艺虽然技术成熟,却存在严重的污染问题,副产大量的废三氯化铝、废硫酸和废水,能耗物耗高,无法满足当前绿色化工的要求。因此,烷基蒽醌的制备技术亟待更新。
3.另外,随着过氧化氢需求量剧增,对蒽醌载体也提出了更高的要求。通过优化烷基蒽醌的溶解性能和化学稳定性来提升过氧化氢产能已成为共识。复合型蒽醌(混合蒽醌)与单一型蒽醌相比,具有更高的溶解度。通过设计和优化蒽醌产品结构,可以开发具有更高溶解性和加氢稳定性的蒽醌产品。新型高效复合型烷基蒽醌产品发展潜力巨大,但相关的产品设计和制备技术均未见广泛报道。
4.us4404140、us6399795、us2002/0035280、us4305879、cn107746372、cn 111068650和cn 110935486分别公开了制备单一型烷基蒽醌产品的方法。
5.jp2010105942公开了一种苯酐法制备混合蒽醌的方法,但产物仍以戊基蒽醌为主。反应中,戊基发生了分解和异构,生成了蒽醌和低碳数的乙基/丁基蒽醌。其中戊基蒽醌含量为98.2-99.1重量%,其余不足3重量%的物质包括蒽醌、乙基蒽醌和丁基蒽醌。
6.us5354937公开了一种通过萘醌法制备2-戊基蒽醌的工艺,基本原理与萘醌法制备蒽醌过程相似,只是额外增加了关键原料2-叔戊基-1,3-丁二烯的制备,该制备工艺较复杂,需通过乙炔与烷基酮经多步反应制得。将制备好的二烯烃与萘醌混合,在压力为0.01-0.5mpa、温度为50~120℃、烯烃与萘醌摩尔比为1:1~1:1.5条件下发生环加成反应生成2-叔戊基四氢蒽醌,而后在强碱的作用下与空气接触发生氧化反应生成2-叔戊基蒽醌,工艺过程总收率为52%。
7.cn107602368a中公开了一种蒽烷基化-氧化法制备2-戊基蒽醌的方法,第一步,蒽与异戊烯发生烷基化反应制备2-戊基蒽,催化剂为mg-mww分子筛;第二步,2-戊基蒽经氧化反应制备2-戊基蒽醌,氧化剂为氧气,催化剂为γ-al2o3负载了mn、mg和fe等制成的固体催化剂,实验结果表明,2-戊基蒽醌的氧化反应收率最高为28.4%。
8.cn 110937988 a中公开了一种通过一步法制备2-烷基蒽醌的工艺,蒽与烷基化试剂、氧化剂一同与特殊催化剂接触后直接制备2-烷基蒽醌。该催化剂成分包括ts-1、ag、so
42-、zr和耐熔无机氧化物,烷基化试剂为c
2-c6的烯烃或醇,氧化剂优选烷基过氧化氢,在固定床反应器内实现一步反应,压力为0.1-10mpa、温度为50-150、体积空速为0.1-10h-1
、气剂比为50:1-1000:1、烷基化试剂与蒽的摩尔比为1-1.5、氧化剂与烷基蒽的摩尔比为1.5-2.5。蒽的转化率为96.1%-99.6%,2-烷基蒽醌选择性为91.5%-96.5%。
9.综上可知,目前尚无复合型烷基蒽醌产品及可行的制备技术报道。
技术实现要素:
10.本发明的目的是提供一种烷基蒽醌组合物,该烷基蒽醌组合物为高碳数多烷基取代的蒽醌组合物,是制备过氧化氢的核心原料,与单一蒽醌产品相比,具有更加优异的溶解性能和反应性能;进一步地,本发明还提出了一种通过蒽制备烷基蒽醌组合物的方法。
11.为了实现上述目的,本发明一方面提供一种烷基蒽醌组合物,其中,所述烷基蒽醌组合物含有蒽醌的烷基取代物,蒽醌的烷基取代物的分子式为c
14+nh8+2n
o2,7≤n≤12;烷基取代基的位置在蒽醌环的α位和/或β位。
12.优选地,所述蒽醌的烷基取代物如结构式(1)所示:
13.,其中,r1-r4表示取代基,
14.任意三个取代基为h,剩余取代基为碳原子数为7-10的烷基取代基;或者,
15.任意两个取代基为h,其余两个取代基各自独立地为碳原子数为2-10的烷基取代基,且碳原子数总和大于或等于7且小于或等于12;或者,
16.任意一个取代基为h,其余的取代基各自独立地为碳原子数为2-8的烷基,且碳原子数总和大于或等于7且小于或等于12。
17.优选地,所述烷基蒽醌组合物含有c
21h22
o2、c
22h24
o2、c
23h26
o2、c
24h28
o2、c
25h30
o2和c
26h32
o2中的至少任意两种物质。
18.本发明第二方面提供一种烷基蒽醌组合物的制备方法,其中,所述制备方法包括:在氧化条件下以及在氧化反应溶剂和氧化催化剂的存在下,将烷基蒽组合物与氧化剂接触进行氧化反应,得到含有烷基蒽醌组合物的烷基蒽氧化产物;所述烷基蒽醌组合物含有蒽醌的烷基取代物,蒽醌的烷基取代物的分子式为c
14+nh8+2n
o2,7≤n≤12;烷基取代基的位置在蒽醌环的α位和/或β位;
19.其中,所述烷基蒽组合物含有蒽的烷基取代物,蒽的烷基取代物的分子式为c
14+nh10+2n
,7≤n≤12;烷基取代基的位置在蒽环的α位和/或β位。
20.优选地,将所述烷基蒽组合物与氧化剂接触的方式为:从蒽烷基化反应产物中分离所述烷基蒽组合物,将分离得到的烷基蒽组合物、氧化催化剂和氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到烷基蒽醌组合物。
21.优选地,将所述烷基蒽组合物与氧化剂接触的方式为:将含有所述烷基蒽组合物的蒽烷基化反应产物、氧化催化剂和选择性含有的氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物,并从所述烷基蒽氧化产物中分离所述烷基蒽醌组合物。
22.优选地:
23.方式1c:蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂相同,将蒽烷基化反应产物中的烷基化催化剂分离,得到含有沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物,所述烷基蒽物系含有所述烷基蒽组合物,
将烷基化产物混合物和氧化催化剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物;
24.方式2c:蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂不同,将蒽烷基化反应产物中的烷基化催化剂和烷基化反应溶剂分离,得到含有沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物,所述烷基蒽物系含有所述烷基蒽组合物,将烷基化产物混合物、氧化催化剂和氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物。
25.本发明提供的烷基蒽醌组合物,是制备过氧化氢的新型高效复合载体,与单一蒽醌相比,具有更高的溶解度和化学稳定性,丰富了“载体”蒽醌的产品类型,是制备过氧化氢的优异载体,有望成为行业未来发展的方向。尤其是本发明提供的烷基蒽醌组合物,以蒽醌的高碳数多烷基取代物为主,与低碳数的单/混合烷基蒽醌相比,具有更高的溶解度和更稳定的反应性能。
26.本发明提供的蒽制备烷基蒽醌组合物的制备方法,可通过调控和匹配反应-分离工艺,实现不同组成、不同结构的烷基蒽醌组合物的制备。本发明通过调控反应来控制产物组成,再匹配分离来控制产品纯度,工艺的有效耦合即可实现烷基蒽醌组合物的定向制备。本发明提供的蒽制备烷基蒽醌组合物的制备工艺灵活性强,适应性广,可满足不同规格的产品需求。此外,本发明提供的制备方法还具有工艺过程简单、高效、污染小的优点。
27.特别是,本发明提供的蒽制备烷基蒽醌组合物的制备方法中,蒽与烷基蒽物系,或蒽醌与烷基蒽醌物系的分离方法,通过在传统蒸馏过程中引入蒸馏溶剂,采用辅助蒸馏技术,可显著提高蒽或蒽醌的分离效率,极大地降低了蒽或蒽醌分离过程的难度,提高整体分离效率。
28.同时,本发明提供的蒽制备烷基蒽醌组合物的制备方法中,烷基蒽氧化方法可以采用两种方式进行,灵活高效,催化剂分离回收难度低,且不存在腐蚀性,降低了设备投资及氧化废液后处理成本。
附图说明
29.图1是本发明提供的一种具体实施方式的由蒽制备烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)的方法的流程图;
30.图2是本发明提供的一种具体实施方式的由蒽制备烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)的方法的流程图;
31.图3是本发明提供的一种具体实施方式的蒸馏溶剂辅助分离蒽的方法的流程图;
32.图4是本发明提供的一种具体实施方式的蒸馏溶剂辅助分离蒽醌的方法的流程图;
33.图5是本发明提供的一种具体实施方式的蒸馏方式1a和1b的方法的流程图;
34.图6是本发明提供的一种具体实施方式的蒸馏方式2a和2b的方法的流程图;
35.图7是本发明提供的一种具体实施方式的蒸馏方式3a和3b的方法的流程图;
36.图8是本发明提供的一种具体实施方式的蒸馏方式4a和4b的方法的流程图。
的含量为1-20重量%,c
25h30
o2的含量为1-15重量%,c
26h32
o2的含量为5-20重量%。
48.根据本发明,基于结构和组成特点,第三种具体实施方式的烷基蒽醌组合物——烷基蒽醌组合物(ⅲ),以所述烷基蒽醌组合物的总重量为基准,c
21h22
o2的含量为0-20重量%,c
22h24
o2的含量为1-97重量%,c
23h26
o2的含量为1-50重量%,c
24h28
o2的含量为1-50重量%,c
25h30
o2的含量为1-50重量%,c
26h32
o2的含量为0-50重量%;优选地,c
21h22
o2的含量为0-10重量%,c
22h24
o2的含量为20-70重量%,c
23h26
o2的含量为1-25重量%,c
24h28
o2的含量为1-25重量%,c
25h30
o2的含量为1-25重量%,c
26h32
o2的含量为0-30重量%;最优选地,c
21h22
o2的含量为0-5重量%,c
22h24
o2的含量为30-70重量%,c
23h26
o2的含量为1-15重量%,c
24h28
o2的含量为1-15重量%,c
25h30
o2的含量为1-20重量%,c
26h32
o2的含量为1-20重量%。
49.根据本发明,基于结构和组成特点,第四种具体实施方式的烷基蒽醌组合物——烷基蒽醌组合物(ⅳ),以所述烷基蒽醌组合物的总重量为基准,c
21h22
o2的含量为0-20重量%,c
22h24
o2的含量为0-50重量%,c
23h26
o2的含量为1-70重量%,c
24h28
o2的含量为1-99重量%,c
25h30
o2的含量为0-40重量%,c
26h32
o2的含量为0-10重量%;优选地,c
21h22
o2的含量为0-10重量%,c
22h24
o2的含量为1-30重量%,c
23h26
o2的含量为10-55重量%,c
24h28
o2的含量为20-70重量%,c
25h30
o2的含量为0-20重量%,c
26h32
o2的含量为0-5重量%;最选地,c
21h22
o2的含量为0-10重量%,c
22h24
o2的含量为1-20重量%,c
23h26
o2的含量为10-50重量%,c
24h28
o2的含量为25-60重量%,c
25h30
o2的含量为0-20重量%,c
26h32
o2的含量为0-5重量%。
50.根据本发明,基于结构和组成特点,第五种具体实施方式的烷基蒽醌组合物——烷基蒽醌组合物(
ⅴ
),以所述烷基蒽醌组合物的总重量为基准,c
21h22
o2的含量为0-20重量%,c
22h24
o2的含量为0-50重量%,c
23h26
o2的含量为0-50重量%,c
24h28
o2的含量为1-70重量%,c
25h30
o2的含量为0-40重量%,c
26h32
o2的含量为1-99重量%;优选地,c
21h22
o2的含量为0-10重量%,c
22h24
o2的含量为1-30重量%,c
23h26
o2的含量为1-30重量%,c
24h28
o2的含量为5-55重量%,c
25h30
o2的含量为0-20重量%,c
26h32
o2的含量为20-70重量%;最优选地,c
21h22
o2的含量为0-5重量%,c
22h24
o2的含量为1-20重量%,c
23h26
o2的含量为1-20重量%,c
24h28
o2的含量为5-20重量%,c
25h30
o2的含量为0-20重量%,c
26h32
o2的含量为35-70重量%。
51.根据本发明,优选情况下,
52.c
21h22
o2为9,10-蒽醌母核与1个庚基相连,或与1个乙基和1个戊基相连,或与1个丙基和1个丁基相连,或与2个乙基和1个丙基相连;
53.c
22h24
o2为9,10-蒽醌母核与1个辛基相连,或与1个乙基和1个己基相连,或与1个丙基和1个戊基相连,或与2个丁基相连,或与2个乙基和1个丁基相连,或与2个丙基和1个乙基相连;
54.c
23h26
o2为9,10-蒽醌母核与1个壬基相连,或与1个乙基和1个庚基相连,或与1个丙基和1个己基相连,或与1个丁基和1个戊基相连,或与2个乙基和1个戊基相连,或与1个乙基、1个丙基和1个丁基相连,或与3个丙基相连;
55.c
24h28
o2为9,10-蒽醌母核与1个癸基相连,或与1个乙基和1个辛基相连,或与1个丙基和1个庚基相连,或与1个丁基和1个己基相连,或与2个戊基相连,或与2个乙基和1个己基相连,或与1个乙基、1个丙基和1个戊基相连,或与1个乙基和2个丁基相连,或与2个丙基和1个丁基相连;
56.c
25h30
o2为9,10-蒽醌母核与1个乙基和1个壬基相连,或与1个丙基和1个辛基相连,
或与1个丁基和1个庚基相连,或与1个戊基和1个己基相连,或与2个乙基和1个庚基相连,或与1个乙基、1个丙基和1个己基相连,或与1个乙基、1个丁基和1个戊基相连,或与2个丙基和1个戊基相连,或与2个丁基和1个丙基相连;
57.c
26h32
o2为9,10-蒽醌母核与1个乙基和1个癸基相连,或与1个丙基和1个壬基相连,或与1个丁基和1个辛基相连,或与1个戊基和1个庚基相连,或与2个己基相连,或与2个乙基和1个辛基相连,或与1个乙基、1个丙基和1个庚基相连,或与1个乙基、1个丁基和1个己基相连,或与1个乙基和2个戊基相连,或与2个丙基和1个己基相连,或与1个丙基、1个丁基和1个戊基相连,或与3个丁基相连。
58.根据本发明,蒽醌的烷基取代物中的烷基取代基选自乙基、丙基、丁基、戊基、己基、庚基及其它们的异构体中的一种或多种;
59.进一步优选地,所述烷基取代基选自乙基、正丙基、异丙基、正丁基、1-甲基丙基、2-甲基丙基、叔丁基、正戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、叔戊基、2,2-二甲基丙基、1,2-二甲基丙基、1-乙基丙基、正己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基、1-乙基丁基、1,1-二甲基-2-甲基丙基、1-甲基-2,2-二甲基丙基、1-甲基-1-乙基丙基、1-乙基-2-甲基丙基和1,1-二甲基戊基中的一种或多种;最优选地,所述烷基取代基选自乙基、异丙基、1-甲基丙基、叔丁基、1-甲基丁基、叔戊基、1-乙基丙基、1,2-二甲基丙基、1-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、1-乙基丁基、1,1-二甲基-2-甲基丙基、1-甲基-2,2-二甲基丙基、1-甲基-1-乙基丙基、1-乙基-2-甲基丙基和1,1-二甲基戊基中的一种或多种。
60.根据本发明,优选情况下,含有2-3个碳原子的烷基取代基的位置在蒽醌环的α位和/或β位,进一步优选在蒽醌环的β位;含有4-7个碳原子的烷基取代基的位置优选在蒽醌环的β位。
61.根据本发明所述的烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)中允许有少量杂质蒽醌和/或低碳数烷基蒽醌c
14+nh8+2n
o2(2≤n≤6)中的一种或多种的存在,而并不影响本发明所述烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)的性质和后续纯化以及应用。其中,以蒽醌、低碳数烷基蒽醌c
14+nh8+2n
o2(2≤n≤6)和本发明所述烷基蒽醌组合物c
14+nh8+2n
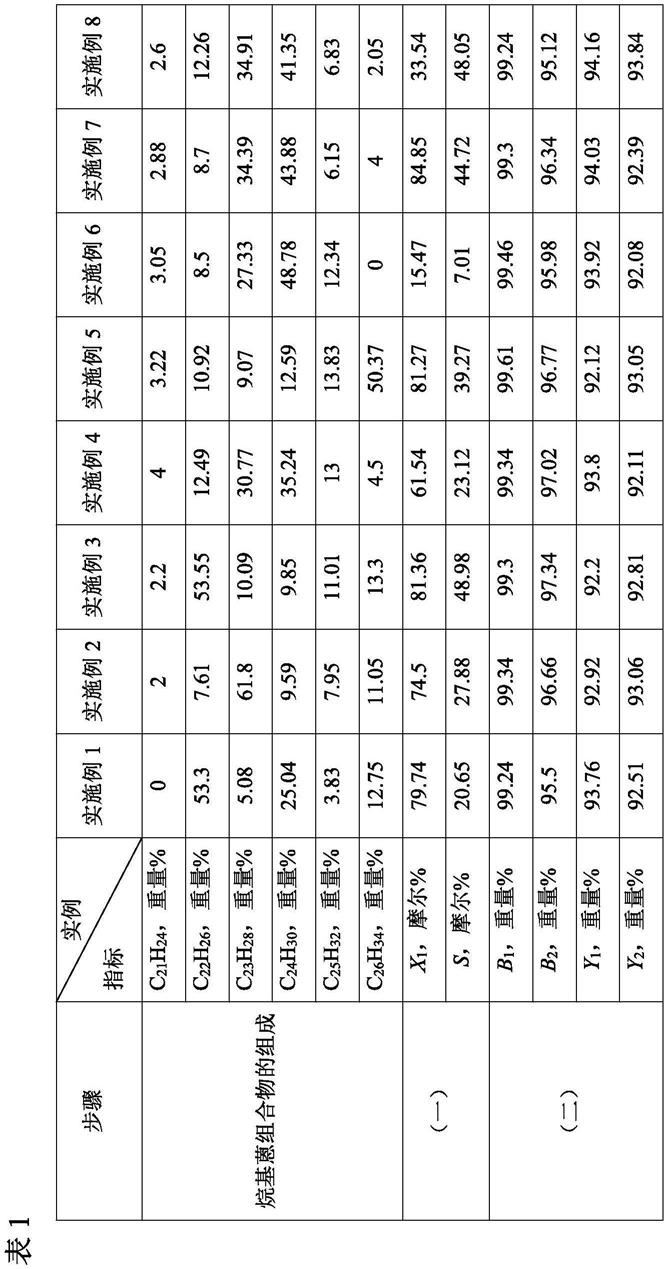
o2(7≤n≤12)的总重量为基准,本发明所述烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)的含量≥90重量%,更优选≥95重量%,进一步优选≥98重量%。
62.根据本发明,本发明所述的烷基蒽醌组合物,特别是根据本发明所述具体实施方式的烷基蒽醌组合物(ⅰ)-(
ⅴ
)的制备方法包括:
63.在氧化条件下以及在氧化反应溶剂和氧化催化剂的存在下,将烷基蒽组合物与氧化剂接触进行氧化反应,得到含有烷基蒽醌组合物的烷基蒽氧化产物;所述烷基蒽醌组合物含有蒽醌的烷基取代物,蒽醌的烷基取代物的分子式为c
14+nh8+2n
o2,7≤n≤12;烷基取代基的位置在蒽醌环的α位和/或β位,优选在β位;
64.其中,所述烷基蒽组合物含有蒽的烷基取代物,蒽的烷基取代物的分子式为c
14+nh10+2n
,7≤n≤12;烷基取代基的位置在蒽环的α位和/或β位,优选在β位。
65.根据本发明,欲制备本发明所述的烷基蒽醌组合物,特别是根据本发明具体实施方式的烷基蒽醌组合物(ⅰ)-(
ⅴ
),可以通过两种方式实现。
66.如图1所示,方式(一):先通过蒽烷基化反应、分离所述烷基蒽组合物c
14+nh10+2n
,7≤n≤12,特别是所述烷基蒽组合物(ⅰ)-(
ⅴ
),再将所述烷基蒽组合物经氧化制备烷基蒽醌组合物,特别是所述烷基蒽醌组合物(ⅰ)-(
ⅴ
)。
67.如图2所示,方式(二):先通过蒽烷基化反应制备烷基蒽物系,将烷基蒽物系经氧化制备烷基蒽醌物系,从烷基蒽醌物系中分离本发明所述的烷基蒽醌组合物,特别是所述烷基蒽醌组合物(ⅰ)-(
ⅴ
)。
68.根据本发明,欲通过方式(一)实现制备本发明所述的烷基蒽醌组合物,特别是所述烷基蒽醌组合物(ⅰ)-(
ⅴ
),则需要先制备用于氧化制备烷基蒽醌组合物的原料——烷基蒽组合物c
14+nh10+2n
,7≤n≤12,特别是所述烷基蒽组合物(ⅰ)-(
ⅴ
)。
69.根据本发明,所述烷基蒽组合物的制备方法包括:在烷基化条件下以及在烷基化反应溶剂和烷基化催化剂的存在下,将蒽与烷基化试剂接触进行烷基化反应,得到含有烷基蒽物系的蒽烷基化反应产物,所述烷基蒽物系含有烷基蒽组合物。
70.根据本发明,所述烷基蒽组合物含有蒽的烷基取代物,所述蒽的烷基取代物的分子式可统一描述为c
14+nh10+2n
(7≤n≤12)。烷基蒽组合物中的每一种物质的分子式均可用c
14+nh10+2n
(7≤n≤12)来表示。烷基蒽组合物中每一种物质的结构均由母核蒽环与烷基取代基构成。烷基取代的位置控制在蒽环的α位和β位,更优选控制在β位。
71.具体来说,所述烷基蒽组合物含有蒽的烷基取代物,蒽的烷基取代物如结构式(2)所示:
72.,其中,r1-r4表示取代基,
73.任意三个取代基为h,剩余取代基为碳原子数为7-10的烷基取代基;或者,
74.任意两个取代基为h,其余两个取代基各自独立地为碳原子数为2-10的烷基取代基,且碳原子数总和大于或等于7且小于或等于12;或者,
75.任意一个取代基为h,其余的取代基各自独立地为碳原子数为2-8的烷基,且碳原子数总和大于或等于7且小于或等于12。
76.根据本发明,优选情况下,所述烷基蒽组合物含有c
21h24
、c
22h26
、c
23h28
、c
24h30
、c
25h32
和c
26h34
中的至少任意两种物质,其中,所述任意两种物质指的是任意两种不同分子式的物质。分子式相同的物质包括多种异构体,包括取代基的个数不同、取代基的结构不同和取代基的位置不同,每种分子式表示具有这种分子式的所有异构体的加和。
77.根据本发明,将蒽与烷基化试剂和催化剂接触的方式可以为各种能够实现蒽的烷基化制备得到烷基蒽组合物的方式。优选地,为了反应更为充分,所述接触的方式为:将含有蒽、烷基化催化剂和烷基化反应溶剂的原料液与烷基化试剂接触进行烷基化反应。
78.根据本发明,将含有蒽、烷基化催化剂和烷基化反应溶剂的原料液与烷基化试剂接触进行烷基化反应的场所,可以是任意一种接触混合良好的反应器,例如,包括釜式反应器和管式反应器,具体可以选自搅拌釜、固定床、移动床、流化床、超重力反应器、微尺度反应器和膜反应器中的一种或多种组合。
79.根据本发明,所述蒽烷基化反应的设备和方法可以按照本领域常规的方式进行。
80.根据本发明,所述烷基化试剂的种类可以参考本领域常规的烷基化试剂,只要能
够满足烷基取代基的总碳数符合本发明的要求即可,例如,所述烷基化试剂可以为含有2-6个碳原子的烷基化试剂中的一种或多种;优选地,所述烷基化试剂为含有2-6个碳原子的烯烃、醇、卤代烃以及醚类物质中的一种或多种;更优选为含有2-6个碳原子的单烯烃、一元醇和一元卤代烃,进一步优选为含有2-6个碳原子的单烯烃。
81.根据本发明,蒽烷基化反应过程中,所述烷基化试剂的用量以能够实现将烷基引入蒽环以制备烷基蒽为准,优选情况下,蒽与烷基化试剂的摩尔比为0.05:1-20:1,优选为0.1:1-5:1。
82.根据本发明,蒽烷基化反应过程中,所述烷基化反应溶剂为能够溶解蒽的惰性有机溶剂。具体来说,所述烷基化反应溶剂为20℃时介电常数为1-10的溶剂,所述烷基化反应溶剂为c6及以上,优选为c
6-c
12
的链烷烃、环烷烃以及芳香烃中的一种或多种;其中,所述芳香烃为取代或未取代,优选为苯的一元或多元取代物中的一种或多种;更优选为苯的多元取代物中的一种或多种,取代基为c
1-c4的烷基和卤族元素中的一种或多种;进一步优选,所述烷基化反应溶剂为苯的多烷基取代物中的一种或多种;最优选,所述烷基化反应溶剂选自1,2,3-三甲苯、1,2,4-三甲苯、1,3,5-三甲苯、1,2,3,5-四甲基苯、1,2,4,5-四甲基苯和1,2,3,4-四甲基苯中的一种或多种。烷基化反应溶剂的用量只要保证蒽能够充分溶解,以达到提供良好的反应介质的作用即可。优选情况下,以蒽和烷基化反应溶剂的总重量为基准,蒽的含量为5-60重量%,优选为8-50重量%。
83.根据本发明,蒽烷基化反应过程中,在烷基化条件下以及在烷基化反应溶剂和催化剂的存在下,将蒽与烷基化试剂接触的方式没有特别限定,优选情况下,为了能够保证烷基化反应更好的进行,所述接触的方式为:将含有蒽、烷基化催化剂和烷基化反应溶剂的原料液与烷基化试剂接触进行烷基化反应。具体来说,先将蒽和催化剂以及烷基化反应溶剂配置成蒽-烷基化催化剂-烷基化反应溶剂的原料液,而后再加入烷基化试剂进行烷基化反应。优选地,蒽-烷基化催化剂-烷基化反应溶剂的原料液的配制温度为80-250℃,更优选为90-200℃。
84.根据本发明,蒽烷基化反应过程中,烷基化反应发生条件一般包括:反应温度可以为80-250℃,优选为90-200℃;反应压力可以为0-2mpa,优选为0-1mpa;反应时间可以为0.01-48h,优选为0.5-24h。
85.根据本发明,蒽烷基化反应过程中,为了使得所述烷基化反应能够更容易进行,所述烷基化反应在烷基化催化剂的存在下进行。具体来说,所述烷基化催化剂可以为能够催化蒽发生烷基化反应的任何形式和种类的催化剂,包括但不限于:高岭土、膨润土、蒙脱土、沸石、x分子筛、y分子筛、β分子筛、mcm-41、sba-15、阳离子交换树脂、全氟磺酸树脂、固载化的硫酸、固载化的磺酸、固载化的磷酸、硅铝复合氧化物、硫酸、高氯酸、四氟硼酸、甲磺酸、苯磺酸、对甲苯磺酸、三氟甲磺酸、三氟化硼、三氯化铝和二氯化锌中的一种或多种;进一步优选自沸石、y分子筛、mcm-41、sba-15、全氟磺酸树脂、固载化的磺酸、硅铝复合氧化物、硫酸、四氟硼酸、甲磺酸、苯磺酸、对甲苯磺酸和三氟甲磺酸中的一种或多种。所述烷基化催化剂的用量亦可以参考本领域的常规用量,以含有蒽、烷基化反应溶剂和烷基化催化剂的原料液的总重量为基准,烷基化催化剂的含量可以为0.01-50重量%,优选为0.5-30重量%,更优选为1-20重量%。
86.根据本发明,具体来说,将蒽与含有2个碳原子的烷基化试剂接触进行烷基化反
应,得到的蒽烷基化反应产物中,含有烷基蒽组合物(ⅰ),其中,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-10重量%,c
22h26
的含量为1-92重量%,c
23h28
的含量为1-20重量%,c
24h30
的含量为5-50重量%,c
25h32
的含量为1-20重量%,c
26h34
的含量为1-50重量%;优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-5重量%,c
22h26
的含量为20-70重量%,c
23h28
的含量为2-10重量%,c
24h30
的含量为10-30重量%,c
25h32
的含量为1-10重量%,c
26h34
的含量为1-30重量%;最优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-2重量%,c
22h26
的含量为35-70重量%,c
23h28
的含量为2-10重量%,c
24h30
的含量为10-30重量%,c
25h32
的含量为1-10重量%,c
26h34
的含量为1-20重量%。
87.具体来说,将蒽与含有3个碳原子的烷基化试剂接触进行烷基化反应,得到的蒽烷基化反应产物中,含有烷基蒽组合物(ⅱ),其中,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-10重量%,c
22h26
的含量为1-20重量%,c
23h28
的含量为1-96重量%,c
24h30
的含量为1-50重量%,c
25h32
的含量为1-50重量%,c
26h34
的含量为1-50重量%;优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-5重量%,c
22h26
的含量为1-10重量%,c
23h28
的含量为20-80重量%,c
24h30
的含量为1-25重量%,c
25h32
的含量为1-25重量%,c
26h34
的含量为1-30重量%;最优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-5重量%,c
22h26
的含量为1-10重量%,c
23h28
的含量为45-75重量%,c
24h30
的含量为1-20重量%,c
25h32
的含量为1-15重量%,c
26h34
的含量为5-20重量%。
88.具体来说,其中,将蒽与含有4个碳原子的烷基化试剂接触进行烷基化反应,得到的蒽烷基化反应产物中,含有烷基蒽组合物(ⅲ),其中,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-20重量%,c
22h26
的含量为1-97重量%,c
23h28
的含量为1-50重量%,c
24h30
的含量为1-50重量%,c
25h32
的含量为1-50重量%,c
26h34
的含量为0-50重量%;优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-10重量%,c
22h26
的含量为20-70重量%,c
23h28
的含量为1-25重量%,c
24h30
的含量为1-25重量%,c
25h32
的含量为1-25重量%,c
26h34
的含量为0-30重量%;最优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-5重量%,c
22h26
的含量为30-70重量%,c
23h28
的含量为1-15重量%,c
24h30
的含量为1-15重量%,c
25h32
的含量为1-20重量%,c
26h34
的含量为1-20重量%。
89.具体来说,其中,将蒽与含有5个碳原子的烷基化试剂接触进行烷基化反应,得到的蒽烷基化反应产物中,含有烷基蒽组合物(ⅳ),其中,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-20重量%,c
22h26
的含量为0-50重量%,c
23h28
的含量为1-70重量%,c
24h30
的含量为1-99重量%,c
25h32
的含量为0-40重量%,c
26h34
的含量为0-10重量%;优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-10重量%,c
22h26
的含量为1-30重量%,c
23h28
的含量为10-55重量%,c
24h30
的含量为20-70重量%,c
25h32
的含量为0-20重量%,c
26h34
的含量为0-5重量%;最优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-10重量%,c
22h26
的含量为1-20重量%,c
23h28
的含量为15-50重量%,c
24h30
的含量为25-60重量%,c
25h32
的含量为0-20重量%,c
26h34
的含量为0-5重量%。
90.具体来说,将蒽与含有6个碳原子的烷基化试剂接触进行烷基化反应,得到的蒽烷基化反应产物中,含有烷基蒽组合物(
ⅴ
),其中,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-20重量%,c
22h26
的含量为0-50重量%,c
23h28
的含量为0-50重量%,c
24h30
的含量为1-70重量%,c
25h32
的含量为0-40重量%,c
26h34
的含量为1-99重量%;优选地,以所
述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-10重量%,c
22h26
的含量为1-30重量%,c
23h28
的含量为1-30重量%,c
24h30
的含量为5-55重量%,c
25h32
的含量为0-20重量%,c
26h34
的含量为20-70重量%;最优选地,以所述烷基蒽组合物的总重量为基准,c
21h24
的含量为0-5重量%,c
22h26
的含量为1-20重量%,c
23h28
的含量为1-20重量%,c
24h30
的含量为5-20重量%,c
25h32
的含量为0-20重量%,c
26h34
的含量为35-70重量%。
91.烷基化试剂在催化剂的作用下形成正碳离子,而正碳离子易于发生二次反应生成多种不同结构的相同碳数或不同碳数的正碳离子;不同正碳离子再与蒽发生烷基化反应,会生成不同取代位置和不同烷基数目及结构的多种烷基蒽产物,但受正碳离子的稳定性和蒽烷基取代产物的稳定性的影响,产物的结构以热力学稳定结构为主,表现出特定的分布和组成。
92.根据本发明,在与不同碳原子数的烷基化试剂反应的过程中,通过控制反应,能够得到不同组成的烷基取代基的烷基蒽组合物(ⅰ)-(
ⅴ
),通过匹配分离来获得烷基蒽组合物(ⅰ)-(
ⅴ
)。基于控制反应调控产品结构特性和组成分布,依靠分离控制产品纯度。通过两步工艺可实现烷基蒽组合物的定向制备。
93.根据本发明,所述的烷基蒽组合物中允许有少量杂质蒽以及低碳数烷基蒽的存在而并不影响所述烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的性质和氧化反应。其中,以蒽、低碳数烷基蒽组合物c
14+nh10+2n
(2≤n≤6)和烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的总重量为基准,烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的含量≥90重量%,更优选≥95重量%,进一步优选≥98重量%。
94.根据本发明,优选情况下,
95.c
21h24
为蒽环与1个庚基相连,或与1个乙基和1个戊基相连,或与1个丙基和1个丁基相连,或与2个乙基和1个丙基相连。
96.c
22h26
为蒽环与1个辛基相连,或与1个乙基和1个己基相连,或与1个丙基和1个戊基相连,或与2个丁基相连,或与2个乙基和1个丁基相连,或与2个丙基和1个乙基相连。
97.c
23h28
为蒽环与1个壬基相连,或与1个乙基和1个庚基相连,或与1个丙基和1个己基相连,或与1个丁基和1个戊基相连,或与2个乙基和1个戊基相连,或与1个乙基、1个丙基和1个丁基相连,或与3个丙基相连。
98.c
24h30
为蒽环与1个癸基相连,或与1个乙基和1个辛基相连,或与1个丙基和1个庚基相连,或与1个丁基和1个己基相连,或与2个戊基相连,或与2个乙基和1个己基相连,或与1个乙基、1个丙基和1个戊基相连,或与1个乙基和2个丁基相连,或与2个丙基和1个丁基相连。
99.c
25h32
为蒽环与1个乙基和1个壬基相连,或与1个丙基和1个辛基相连,或与1个丁基和1个庚基相连,或与1个戊基和1个己基相连,或与2个乙基和1个庚基相连,或与1个乙基、1个丙基和1个己基相连,或与1个乙基、1个丁基和1个戊基相连,或与2个丙基和1个戊基相连,或与2个丁基和1个丙基相连。
100.c
26h34
为蒽环与1个乙基和1个癸基相连,或与1个丙基和1个壬基相连,或与1个丁基和1个辛基相连,或与1个戊基和1个庚基相连,或与2个己基相连,或与2个乙基和1个辛基相连,或与1个乙基、1个丙基和1个庚基相连,或与1个乙基、1个丁基和1个己基相连,或与1个乙基和2个戊基相连,或与2个丙基和1个己基相连,或与1个丙基、1个丁基和1个戊基相
连,或与3个丁基相连。
101.根据本发明,蒽的烷基取代物中的烷基取代基可以选自乙基、丙基、丁基、戊基、己基和庚基中的一种或多种;优选地,所述烷基取代基选自乙基、正丙基、异丙基、正丁基、1-甲基丙基、2-甲基丙基、叔丁基、正戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、叔戊基、2,2-二甲基丙基、1,2-二甲基丙基、1-乙基丙基、正己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基、1-乙基丁基、1,1-二甲基-2-甲基丙基、1-甲基-2,2-二甲基丙基、1-甲基-1-乙基丙基、1-乙基-2-甲基丙基和1,1-二甲基戊基中中的一种或多种;更优选地,所述烷基取代基选自乙基、异丙基、1-甲基丙基、叔丁基、1-甲基丁基、叔戊基、1-乙基丙基、1,2-二甲基丙基、1-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、1-乙基丁基、1,1-二甲基-2-甲基丙基、1-甲基-2,2-二甲基丙基、1-甲基-1-乙基丙基、1-乙基-2-甲基丙基和1,1-二甲基戊基中的一种或多种。
102.根据本发明,优选情况下,含有2-3个碳原子的烷基取代基的位置在蒽环的α位和/或β位,进一步优选在蒽环的β位;含有4-7个碳原子的烷基取代基的位置优选在蒽环的β位。
103.根据本发明,根据方式(一),如图1所示,将所述烷基蒽组合物与氧化剂接触的方式为:从蒽烷基化反应产物中分离所述烷基蒽组合物c
14+nh10+2n
(7≤n≤12),将分离得到的烷基蒽组合物、氧化催化剂和氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)。蒽烷基化反应产物含有沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系,所述烷基蒽物系含有所述烷基蒽组合物。因此,所述方法还包括从蒽烷基化反应产物中分离所述烷基蒽组合物。
104.方式1:
105.烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量小于或等于1重量%,优选地,烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量小于或等于5重量%;
106.所述分离方法包括:
107.预分离:分离沸点低于蒽的轻组分,得到烷基蒽物系;
108.烷基蒽组合物的分离:通过蒸馏从含有烷基蒽的物系中分离出所述烷基蒽组合物c
14+nh10+2n
,7≤n≤12;或者,
109.方式2:
110.烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量高于或等于10重量%,优选地,烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量高于5重量%;
111.所述分离方法包括:
112.预分离:分离沸点低于蒽的轻组分,得到含有蒽和烷基蒽物系的混合物;
113.蒸馏溶剂辅助分离蒽:在蒸馏溶剂的存在下,将含有蒽和烷基蒽物系的混合物进行蒸馏,并收集烷基蒽物系,所述蒸馏溶剂为在辅助分离蒽的过程中能够溶解蒽的、沸点介于100-340℃的有机溶剂;
114.烷基蒽组合物的分离:通过蒸馏从烷基蒽物系中分离所述烷基蒽组合物c
14+nh10+2n
,7≤n≤12。
115.烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量大于1重量%且小于10重量%,采用方式1或方式2中的任意一种分离方式,优选地,烷基化反应产物中沸点≥蒽的
沸点的混合物内,蒽的含量小于或等于5重量%,采用方式1的分离方式;烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量高于5重量%,采用方式2的分离方式。
116.根据本发明,由原料蒽经烷基化反应制备烷基蒽组合物,所获得的蒽烷基化反应产物混合物中含有反应溶剂、烷基化催化剂、剩余的蒽、烷基蒽物系和其他副产物。本领域技术人员公知的是,由于反应方法和条件不同,若蒽可全部转化,则蒽烷基化反应产物混合物中不含蒽;若蒽不能全部转化,则蒽烷基化反应产物混合物中含有部分剩余的蒽。若烷基化试剂可以全部转化,但未全部转化为烷基蒽,则蒽烷基化反应产物混合物中含有烷基化试剂的副反应产物;若烷基化试剂不能全部转化,且未全部转化为烷基蒽,则蒽烷基化反应产物混合物中含有烷基化试剂和烷基化试剂的副反应产物。蒽烷基化反应产物中,反应溶剂、烷基化试剂和烷基化试剂副反应产物的沸点均低于蒽的沸点,因此,把蒽烷基化反应产物中沸点低于蒽的物质统称为轻组分。
117.根据本发明的方式1,若烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量小于或等于1重量%,优选地,烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量小于或等于5重量%;则可在先分离轻组分之后直接从烷基蒽物系中分离所述烷基蒽组合物,少量的蒽杂质不影响所述烷基蒽组合物的性质,所述分离方法包括:
118.预分离:分离沸点低于蒽的轻组分,得到烷基蒽物系;
119.烷基蒽组合物的分离:通过蒸馏从含有烷基蒽的物系中分离出所述烷基蒽组合物c
14+nh10+2n
,7≤n≤12。
120.根据本发明,在蒽烷基化反应过程中,由于反应特性及方法决定,方式1分离轻组分和优选先分离烷基化催化剂后,会获得多种烷基蒽的混合物,即烷基蒽物系,所述烷基蒽物系中含有本发明需要的烷基蒽组合物c
14+nh10+2n
(7≤n≤12)、沸点小于c
21h24
的沸点的物质,如其他烷基蒽产物c
14+nh10+2n
(2≤n≤6)、其他烷基蒽副产物和其他高沸点副产物。因此,需要进一步通过减压蒸馏的方法,从烷基蒽物系中分离出所述烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。
121.根据本发明的方式1中,如图5所示,烷基蒽组合物的分离步骤中,通过蒸馏从烷基蒽物系中分离出所述烷基蒽组合物c
14+nh10+2n
,7≤n≤12的方法包括:
122.方式1a:
123.在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量小于或等于1重量%,优选地,在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量小于或等于5重量%;从含有烷基蒽的物系中蒸馏分离出烷基蒽组合物c
14+nh10+2n
,7≤n≤12的蒸馏条件包括:塔顶压力为0.005-20kpa,塔底温度为200-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为210-400℃,理论板数为30-75,塔顶回流比为1-7;或者,
124.方式1b:
125.在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量高于或等于10重量%,优选地,在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量高于5重量%;先从烷基蒽物系中蒸馏分离出沸点低于c
21h24
的沸点的物质,蒸馏条件包括:蒸馏塔顶压力为0.01-20kpa,塔底温度为200-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为210-400℃,理论板数为30-75,塔顶回流比为1-7;从除去
沸点低于c
21h24
的物质的烷基蒽物系中蒸馏分离出烷基蒽组合物c
14+nh10+2n
,7≤n≤12的蒸馏条件包括:蒸馏塔顶压力为0.005-20kpa,塔底温度为200-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为210-400℃,理论板数为30-75,塔顶回流比为1-7。
126.在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量大于1重量%且小于10重量%,采用方式1a或方式1b中的任意一种分离方式,优选地,在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量小于或等于5重量%,采用方式1a的分离方式;在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量高于5重量%,采用方式1b的分离方式。
127.根据本发明的方式2,若烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量高于或等于10重量%,优选地,烷基化反应产物中沸点≥蒽的沸点的混合物内,蒽的含量高于5重量%,在分离轻组分之后,则需要先将蒽分离除去,然后再分离所述烷基蒽组合物。
128.根据物性分析可知,蒽的沸点为340℃,烷基蒽产物与蒽属同系物,彼此间存在沸点差异,可通过减压蒸馏技术来实现产物分离。但技术难点在于,蒽的熔点高达215℃,单独采用减压蒸馏技术来分离高熔点的蒽,操作难度大,管路极易发生堵塞问题,严重影响工艺的连续稳定运行。另外,蒽极易升华,升华过程难以控制,管路发生堵塞的机会显著增加。因此,单纯采用减压蒸馏技术来实现蒽-烷基蒽物系的分离是不切实际的。
129.因此,本发明的发明人提出溶剂辅助分离蒽和蒸馏分离烷基蒽物系混合物的方法。烷基蒽由于侧链取代基团的存在,破坏了蒽环结构的高度规整性,使得烷基蒽产物熔点明显降低,降低了后续蒸馏分离的难度。为此,本发明的发明人提出先采用溶剂辅助蒸馏技术,将熔点最高且最难实现分离操作的蒽分离除去,而后根据沸点差异,采用蒸馏的方法分离烷基蒽组合物以实现进一步分离。
130.根据本发明的一种具体实施方式,如图1和图3所示,蒸馏溶剂辅助分离蒽在蒸馏塔内进行。具体来说,预分离后,将含有蒽和烷基蒽物系的混合物引入蒸馏塔,该蒸馏过程可以是间歇式的,也可以是连续式的。蒸馏时,向蒸馏塔引入蒸馏溶剂,蒽在蒸馏条件下开始逐渐蒸出,同时引入的蒸馏溶剂进入蒸馏塔后也开始大量气化,并且与蒽一同蒸出进入塔顶冷凝器内进行冷凝。在大量的气化和液化的蒸馏溶剂分子氛围下,蒽无法经凝华和凝固结晶,而是溶解在蒸馏溶剂中形成溶液并随之一起流动,进而解决了蒽易堵塞管路的问题。蒸馏溶剂与蒽形成的溶液部分回流进入蒸馏塔重复蒸馏,部分流入塔顶产品罐收集。通过蒸馏溶剂的引入,控制其在塔顶与塔顶冷凝器间循环,同时调控进料位置、温度和用量,使之溶解蒽形成溶液一同顺利采出,即可实现蒽的高效分离,又可解决蒽蒸馏时的高度易凝的难题。
131.因此,根据本发明,在蒸馏溶剂辅助分离蒽过程中,所述蒸馏溶剂为在辅助分离蒽的过程中能够溶解蒽的、沸点介于100-340℃的有机溶剂。
132.优选地,所述蒸馏溶剂为沸点介于200-340℃的有机溶剂,可选自c
12-c
19
的直链烷烃和/或支链烷烃、卤代烃、芳香烃、醇、酮、酯和醚中的一种或多种;更优选地,所述烷烃为c
12-c
17
的直链烷烃和/或支链烷烃中的一种或多种;更优选地,所述卤代烃选自三氯苯、四氯苯、三溴苯、四溴苯、氯代c
10-c
18
烷、溴代c
10-c
18
烷中的一种或多种;更优选地,所述芳香烃为苯的烷基取代物,取代烷基的总碳数为4-12;进一步优选为丁基苯、戊基苯、己基苯、庚基
苯、辛基苯、壬基苯、癸基苯、十一烷基苯、十二烷基苯、三乙基苯、四乙基苯、二丙基苯、三丙基苯、二丁基苯、二戊基苯中的一种或多种;更优选地,所述芳烃烷为苯的取代物,进一步优选为二苯甲烷及其烷基取代物,二苯乙烷及其烷基取代物;更优选为二苯甲烷、甲基二苯甲烷、1,2-二苯乙烷的一种或多种;更优选地,所述芳烃烷为萘或萘的烷基取代物,萘的取代烷基总碳数为1-4;进一步优选萘、甲基萘、二甲基萘、乙基萘、二乙基萘、丙基萘、甲基乙基萘、丁基萘中的一种或多种;更优选地,所述醇选自苯甲醇、丙三醇、二甘醇、三甘醇、四甘醇中的一种或多种;更优选地,所述酮选自1,1,3-三甲基环己烯酮、n-甲基吡咯烷酮、1,3-二甲基-2-咪唑啉酮中的一种或多种;更优选地,所述酯选自二甲酸酯、苯甲酸乙酯、邻苯二甲酸二甲酯、邻苯二甲酸二丁酯、乙二醇碳酸酯、丙二醇碳酸酯、磷酸三辛酯中的一种或多种;更优选地,所述醚选自乙二醇单苯醚、二乙二醇单丁醚、二苯醚和环丁砜中的一种或多种。
133.方式2中,所述蒸馏溶剂辅助分离蒽的条件包括:蒸馏塔顶压力为0.5-40kpa,塔底温度为200-450℃,理论板数为12-55,塔顶回流比为0.1-4;优选地,蒸馏塔顶压力为1-20kpa,塔底温度为230-400℃,理论板数为16-50,塔顶回流比为0.2-1。所述蒸馏溶剂的用量可以根据进行蒸馏的含有蒽和烷基蒽物系的混合物中蒽的含量进行选择,以能够实现充分分离蒽以提高烷基蒽物系纯度为准。优选地,蒸馏溶剂与蒽的质量比为0.1:1-30:1。在确保能够获得令人满意的烷基蒽物系的纯度的条件下,从进一步降低本发明的方法的成本的角度出发,蒸馏溶剂与蒽的质量比为1:1-15:1。
134.根据本发明,如图3所示,在蒸馏溶剂辅助分离蒽过程中,塔顶收集的产品为蒸馏溶剂和蒽的混合物,需要将两者全部或部分的分离。优选情况下,所述蒸馏溶剂辅助分离蒽的步骤中还可以包括:收集含有蒽和蒸馏溶剂的混合物,并将蒽和蒸馏溶剂分离,回收得到蒽,并可以将蒸馏溶剂重复再利用。从蒸馏溶剂和蒽的混合物中分离蒽和蒸馏溶剂可以依据溶解度的差异,采用包括萃取和结晶的方法;也可以依据沸点的差异,采用蒸馏的方法。
135.根据本发明,优选采用蒸馏的方法分离蒸馏溶剂和蒽。所述蒸馏可以采用本领域公知的各种蒸馏设备,例如:筛板塔或者填料塔,更优选填料塔。具体来说,将含有蒽和蒸馏溶剂的混合物进行蒸馏,蒸馏条件包括:塔顶压力为1-100kpa,塔底温度为160-350℃,理论板数为6-40,塔顶回流比为0.1-3;进一步优选,塔顶压力为20-60kpa,塔底温度为200-310℃,理论板数为8-30,塔顶回流比为0.2-2。
136.根据本发明,在蒽烷基化反应过程中,由于反应特性及方法决定,方式2分离轻组分和烷基化催化剂以及除去反应剩余的蒽之后,会获得含有多种烷基蒽的混合物,即烷基蒽物系,所述烷基蒽物系中含有本发明需要的烷基蒽组合物c
14+nh10+2n
(7≤n≤12)、沸点小于c
21h24
的沸点的物质,如其他烷基蒽产物c
14+nh10+2n
(2≤n≤6)、其他烷基蒽副产物和其他高沸点副产物。因此,需要进一步通过减压蒸馏的方法,从烷基蒽物系中分别分离出所述烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。
137.根据本发明的方式2,如图6所示,烷基蒽组合物的分离步骤中,通过蒸馏从烷基蒽物系中分离出所述烷基蒽组合物c
14+nh10+2n
,7≤n≤12的方法包括:
138.方式2a:
139.在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量小于或等于1重量%,优选地,在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量小于或等于5重量%;从烷基蒽物系中蒸馏分离出烷基蒽组合物c
14+nh10+2n
,7≤n≤12的蒸馏条件包括:塔顶
压力为0.005-20kpa,塔底温度为200-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为210-400℃,理论板数为30-75,塔顶回流比为1-7;或者,
140.方式2b:
141.在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量高于或等于10重量%,优选地,在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量高于5重量%;先从烷基蒽物系中蒸馏分离出沸点低于c
21h24
的沸点的物质,蒸馏条件包括:蒸馏塔顶压力为0.01-20kpa,塔底温度为200-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为210-400℃,理论板数为30-75,塔顶回流比为1-7;从除去沸点低于c
21h24
的物质的烷基蒽物系中蒸馏分离出烷基蒽组合物c
14+nh10+2n
,7≤n≤12的蒸馏条件包括:蒸馏塔顶压力为0.005-20kpa,塔底温度为200-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为210-400℃,理论板数为30-75,塔顶回流比为1-7。
142.在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量大于1重量%且小于10重量%,采用方式2a或方式2b中的任意一种分离方式,优选地,在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量小于或等于5重量%,采用方式2a的分离方式;在含有烷基蒽的物系中,沸点小于c
21h24
的沸点的物质的含量高于5重量%,采用方式2b的分离方式。
143.根据本发明,所述蒽烷基化反应产物中除了含有蒽和烷基蒽物系(所述烷基蒽物系含有所述烷基蒽组合物)之外,还含有上步蒽烷基化反应过程中,由于反应方法和操作条件的不同,可能会带入或产生的沸点低于蒽的轻组分以及烷基化催化剂。其中,沸点低于蒽的轻组分含有蒽经烷基化反应制备烷基蒽物系的反应溶剂、烷基化试剂和烷基化反应产生的副产物(例如烷基化反应后剩余的烷基化试剂和烷基化试剂自身发生副反应产生的烷基化试剂副反应产物),统称为轻组分。因此,在所述烷基蒽组合物的制备和分离方法的方式1的烷基蒽组合物的分离之前,或者方式2的蒸馏溶剂辅助分离蒽之前,还包括分离轻组分的步骤即预分离的步骤。
144.根据本发明,所述分离轻组分的方法可以采用本领域常规的分离方法。优选情况下,从进一步提高分离效率以及操作简便的角度考虑,采用常压或减压蒸馏的方法分离含有沸点低于蒽的轻组分、选择性含有的蒽和烷基蒽物系的混合物中的轻组分。
145.根据本发明的一种具体实施方式,从进一步提高分离效率以及操作简便的角度考虑,采用减压蒸馏的方法进行预分离。具体来说,所述预分离的方法包括:将含有沸点低于蒽的轻组分、蒽以及烷基蒽物系(所述烷基蒽物系含有所述烷基蒽组合物)的混合物在蒸馏塔中进行蒸馏,得到含有沸点低于蒽的轻组分的馏出物以及含有蒽和烷基蒽物系的塔底产物,蒸馏的条件包括:蒸馏温度为50-350℃,优选为60-300℃;蒸馏压力为0.1-20kpa,优选0.5-15kpa。此外,可以将分离的反应溶剂按照反应的要求循环使用或者收集处理。
146.根据本发明,由于所述蒽烷基化反应产物中还含有蒽经烷基化反应制备系列烷基蒽产物的烷基化催化剂,因此,为了保证后续步骤的分离效果,优选情况下,所述制备方法还包括在方式1或方式2的预分离之前,先分离烷基化催化剂。所述分离烷基化催化剂的方法可以采用本领域常规的分离方法,例如沉降、过滤和离心分离中的一种或多种。
147.根据本发明,通过蒽的烷基化反应获得的烷基蒽组合物c
14+nh10+2n
(7≤n≤12)为目标产物,如其中仍含有其他杂质,可进一步通过其他常规分离方法或组合分离方法进行提纯,包括蒸馏、萃取和结晶。
148.根据本发明的方式(一),如图1所示,将所述烷基蒽组合物与氧化剂接触的方式为:从蒽烷基化反应产物中分离所述烷基蒽组合物,将分离得到的烷基蒽组合物、氧化催化剂和氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到烷基蒽醌组合物。
149.根据本发明,欲通过方式(二)实现制备本发明所述的烷基蒽醌组合物,特别是烷基蒽醌组合物(ⅰ)-(
ⅴ
),需要先通过蒽烷基化反应制备烷基化反应产物,所述烷基化反应产物中含有本发明所述的烷基蒽组合物c
14+nh10+2n
(7≤n≤12),通过蒽烷基化反应制备烷基化反应产物的方法如上所述,不再赘述。然后将含有所述烷基蒽组合物的蒽烷基化反应产物、氧化催化剂和选择性含有的氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物,并从所述烷基蒽氧化产物中分离所述烷基蒽醌组合物。
150.当蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂不同时,需要将蒽烷基化反应产物中的烷基化催化剂和烷基化反应溶剂分离,得到含有沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物,所述烷基蒽物系含有烷基蒽组合物,将所述分离出烷基化催化剂和烷基化反应溶剂后获得的烷基化产物混合物与氧化催化剂和氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物,再分离出烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)。
151.当蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂相同时,将蒽烷基化反应产物中的烷基化催化剂分离,得到含有沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物,可直接与氧化催化剂混合得到的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物,再分离出烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12),省去了反应溶剂的蒸馏更换步骤。
152.具体来说,根据本发明的方式(二),如图2所示,将所述烷基蒽组合物与氧化剂接触的方式为:
153.将含有所述烷基蒽组合物的蒽烷基化反应产物、氧化催化剂和选择性含有的氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物,并从所述烷基蒽氧化产物中分离所述烷基蒽醌组合物。
154.优选地:
155.方式1c:蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂相同,将蒽烷基化反应产物中的烷基化催化剂分离,得到含有沸点低于蒽的轻组分(此时,该沸点低于蒽的轻组分含有烷基化反应溶剂和其他沸点高于烷基化反应溶剂但低于蒽的副产物)、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物,所述烷基蒽物系含有所述烷基蒽组合物,将烷基化产物混合物和氧化催化剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物;
156.方式2c:蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂不同,将蒽烷基化反应产物中的烷基化催化剂和烷基化反应溶剂分离,得到含有沸点低于蒽的轻组分(此时,该沸点低于蒽的轻组分就是沸点高于烷基化反应溶剂但低于蒽的副产
物)、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物,所述烷基蒽物系含有所述烷基蒽组合物,将烷基化产物混合物、氧化催化剂和氧化反应溶剂的混合物与氧化剂接触进行氧化反应,得到含有所述烷基蒽醌组合物的烷基蒽氧化产物。
157.根据本发明,无论是通过方式(一)制备并分离所述烷基蒽组合物,还是通过方式(二)制备所述烷基蒽物系,均需要经过氧化反应来获得烷基蒽醌产物。
158.根据本发明,所述氧化剂通常为过氧化氢,在氧化过程中,为了便于操作,优选将作为氧化剂的过氧化氢以过氧化氢水溶液的形式使用,过氧化氢水溶液的浓度没有特别限定,可以参考本领域的常规选择。
159.根据本发明,在氧化过程中,所述氧化剂的用量以能够实现将烷基蒽氧化以制备烷基蒽醌为准。
160.具体地,在方式(一)中,氧化剂与所述分离得到的烷基蒽组合物中具有蒽环结构的所有物质总和的摩尔比为0.01:1-100:1,优选为1:1-50:1。
161.具体地,在方式(二)中,氧化剂与烷基化产物混合物(分离烷基化催化剂和选择性分离烷基化反应溶剂后得到含有沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物)中,即沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系的混合物中具有蒽环结构的所有物质总和的摩尔比为0.01:1-100:1,优选为1:1-50:1。
162.根据本发明,所述氧化催化剂选自碱土金属的氧化物、碱土金属的氢氧化物、过渡金属的含氧化合物以及镧系金属的含氧化合物中的一种或多种。优选情况下,在氧化过程中,所述催化剂选自ⅱa族、ⅳb族、
ⅴ
b族、ⅵb族、ⅶb族、
ⅷ
族金属的含氧化合物、镧系金属的含氧化合物中的一种或多种。例如,所述ⅱa族可以为be、mg、ca、sr、ba的含氧化合物,所述ⅳb族可以为ti、zr的含氧化合物,所述
ⅴ
b族可以为v、nb、ta的含氧化合物,所述ⅵb族可以为cr、mo、w的含氧化合物,所述ⅶb族可以为mn、re的含氧化合物,所述
ⅷ
族可以为fe、co、ni、ru、rh、pd、os、ir、pt的含氧化合物,所述镧系可以为la、ce、pr、nd、pm、sm、eu、gd、tb、dy、ho、er、tm、yb、lu的含氧化合物。更优选,所述催化剂选自ca、ba、ti、zr、v、cr、mo、w、mn、ru、co、ni、la和ce的含氧化合物中的一种或多种。最优选,所述催化剂选自氢氧化钙、氢氧化钡、四价钛含氧化物包括偏钛酸、四价锆含氧化物包括二氧化锆和硝酸氧锆、五价钒含氧化物包括偏钒酸钠、六价铬含氧化物包括铬酸钾和三氧化二铬、六价钼含氧化物包括钼酸钠、钼酸铵和三氧化钼、六价钨含氧化物包括钨酸钠、三价锰和四价锰含氧化物包括三氧化二锰和二氧化锰、四价钌含氧化物包括二氧化钌、三价钴含氧化物包括三氧化二钴、二价镍和三价镍含氧化物包括氧化镍和三氧化二镍、三价镧含氧化物包括硝酸镧和三氧化二镧、四价铈含氧化物包括二氧化铈中的一种或多种。
163.根据本发明,进一步优选情况下,将氧化剂过氧化氢与选自碱土金属的氧化物、碱土金属的氢氧化物、过渡金属的含氧化合物以及镧系金属的含氧化合物中的一种或多种的催化剂组合使用,可有效实现烷基蒽的氧化,且氧化体系简单高效,催化剂分离回收难度低,且不存在腐蚀性,降低了设备投资及氧化废液后处理成本。
164.根据本发明,所述氧化过程中,氧化剂与氧化催化剂用量的可选择范围较宽,优选情况下,为了更好的实现本发明的发明目的,氧化剂与氧化催化剂的摩尔比为0.01:1-100:1,更优选为0.1:1-30:1。
165.根据本发明,在氧化过程中,除了上述的过氧化氢氧化剂与特定的催化剂的组合
之外,所述氧化反应的设备、条件和方法可以按照本领域常规的方式进行。
166.根据本发明,将含有烷基蒽组合物、氧化催化剂和氧化反应溶剂的原料液与氧化剂接触进行氧化反应的场所,可以是任意一种接触混合良好的反应器,包括釜式反应器和管式反应器,包括搅拌釜、固定床、移动床、流化床、超重力反应器、微尺度反应器和膜反应器中的任意一种或组合。
167.根据本发明,在氧化过程中,氧化反应发生条件一般包括:反应温度为10-200℃,优选为20-120℃;反应压力为0-1mpa,优选为0-0.5mpa;反应时间为0.01-48h,优选为0.5-24h。
168.根据本发明,氧化过程中,所述氧化反应溶剂为能够溶解烷基蒽的惰性有机溶剂。其中,氧化反应溶剂为20℃时介电常数1-50的溶剂,所述氧化反应溶剂为c6及以上,优选为c
6-c
12
的链烷烃、环烷烃以及芳香烃中的一种或多种;其中,所述芳香烃为取代或未取代,优选为苯的一元、二元或多元取代物中的一种或多种;更优选为苯的多元取代物中的一种或多种,取代基为c
1-c4的烷基和卤族元素中的一种或多种;更优选,所述氧化反应溶剂为苯的多烷基取代物中的一种或多种;更优选,所述氧化反应溶剂选自1,2,3-三甲苯、1,2,4-三甲苯、1,3,5-三甲苯、1,2,3,5-四甲基苯、1,2,4,5-四甲基苯和1,2,3,4-四甲基苯中的一种或多种;更优选地,氧化反应溶剂为碳数为1~4的脂肪醇、四氢呋喃、丙酮、乙酸乙酯、乙腈、二甲基亚砜、环丁砜、n,n-二甲基苯胺、甲酰胺、乙酰胺、n-烷基取代酰胺和n-烷基吡咯烷酮中的一种或多种,其中,烷基取代基的个数为1-2,每个烷基取代基各自独立地为c
1-c4的烷基;最优选,所述氧化反应溶剂选自甲醇、叔丁醇、丙酮、二甲基亚砜、环丁砜、n,n-二甲基苯胺、甲酰胺、乙酰胺、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n,n-二甲基丙酰胺、n-甲基吡咯烷酮和n-乙基吡咯烷酮中的一种或多种。
169.根据本发明,在氧化过程中,所述氧化反应溶剂的用量只要保证烷基蒽能够充分溶解,以达到提供良好的反应介质的作用即可。
170.具体地,在方式(一)中,以所述分离得到的烷基蒽组合物以及氧化反应溶剂的总重量为基准,所述分离得到的烷基蒽组合物的含量为0.1-80重量%,优选为5-50重量%。
171.具体地,在方式(二)中,若蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂相同,以分离烷基化催化剂后剩余的反应液总重量为基准,其中沸点低于蒽的轻组分(不含烷基化反应溶剂)、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物的含量为0.1-80重量%,优选为5-50重量%。
172.具体地,在方式(二)中,若蒽烷基化反应所用烷基化反应溶剂与烷基蒽氧化反应所用氧化反应溶剂不同,分离烷基化催化剂和烷基化反应溶剂后得到含有沸点低于蒽的轻组分、选择性含有的蒽以及烷基蒽物系的烷基化产物混合物以及氧化反应溶剂的总重量为基准,烷基化反应产物混合物的含量为0.1-80重量%,优选为5-50重量%。
173.根据本发明的方式(二),将烷基化反应产物中的烷基化催化剂分离的方法可以参考本领域常规的分离方法,例如沉降、过滤和离心分离中的一种或多种。根据本发明的方式(二),将蒽烷基化反应产物中的烷基化反应溶剂分离的方法可以参考本领域常规的分离方法,例如,采用常压或减压蒸馏的方法分离所述烷基化反应溶剂,在此不再赘述。
174.根据本发明的方式(二),所述烷基蒽氧化产物含有沸点低于蒽醌的物质以及选择性含有的蒽醌和烷基蒽醌物系,所述烷基蒽醌物系含有所述烷基蒽醌组合物,所述方法还
包括从烷基蒽氧化产物中分离所述烷基蒽醌组合物。从所述烷基蒽氧化产物中分离所述烷基蒽醌组合物的方法包括:
175.方式3:
176.烷基蒽氧化产物中沸点大于或等于蒽醌的混合物内,蒽醌的含量小于或等于1重量%;优选地,蒽醌的含量小于或等于5重量%;
177.所述分离方法包括:
178.预分离:分离沸点低于蒽醌的物质,得到烷基蒽醌物系;
179.烷基蒽醌组合物的分离:通过蒸馏从烷基蒽醌物系中分离出所述烷基蒽醌组合物c
14+nh8+2n
o2,7≤n≤12;
180.方式4:
181.烷基蒽氧化产物中沸点大于等于蒽醌的混合物内,蒽醌的含量高于或等于10重量%;优选地,蒽醌的含量高于5重量%;
182.所述分离方法包括:
183.预分离:分离沸点低于蒽醌的物质,得到含有蒽醌和烷基蒽醌物系的混合物;
184.分离蒽醌:所述分离蒽醌的方法选自萃取分离、熔融结晶分离蒽醌、溶剂结晶分离蒽醌、蒸馏分离蒽醌中的一种或多种,优选蒸馏分离蒽醌,进一步优选,采用蒸馏溶剂辅助分离蒽醌的方法:在蒸馏溶剂的存在下,将含有蒽醌和烷基蒽醌物系的混合物进行蒸馏,并收集烷基蒽醌物系,所述蒸馏溶剂为在辅助分离蒽醌的过程中能够溶解蒽醌的、沸点介于100-340℃的有机溶剂;
185.烷基蒽醌组合物的分离:通过蒸馏从烷基蒽醌物系中分离出烷基蒽醌组合物c
14+nh8+2n
o2,7≤n≤12。
186.烷基蒽氧化产物中沸点≥蒽醌的沸点的混合物内,蒽醌的含量大于1重量%且小于10重量%,采用方式3或方式4中的任意一种分离方式,优选地,烷基蒽氧化产物中沸点≥蒽醌的沸点的混合物内,蒽醌的含量小于或等于5重量%,采用方式3的分离方式;烷基蒽氧化产物中沸点≥蒽醌的沸点的混合物内,蒽醌的含量高于5重量%,采用方式4的分离方式。
187.根据本发明,所述烷基蒽氧化产物含有沸点低于蒽醌的物质以及选择性含有的蒽醌和烷基蒽醌物系和其他副产物。由原料蒽经烷基化反应制备蒽烷基化产物,分离其中的烷基化催化剂后得到反应液,其中的烷基蒽物系c
14+nh10+2n
(2≤n≤20)经氧化得到烷基蒽醌物系c
14+nh8+2n
o2(2≤n≤20)。所述反应液中的蒽和烷基蒽物系经氧化后,控制氧化反应条件,尽量将蒽和烷基蒽物系全部或极大部分地转变为蒽醌和烷基蒽醌物系c
14+nh8+2n
o2(2≤n≤20)。
188.根据本发明,方式3中,若在原料蒽经烷基化反应进行蒽烷基化反应过程中,若通过控制反应的方法和条件,使得蒽全部或大部分被转化,烷基蒽氧化产物中沸点大于或等于蒽醌的混合物内,蒽醌的含量小于或等于1重量%;优选地,蒽醌的含量小于或等于5重量%;则可在先分离轻组分之后直接从含有烷基蒽醌的物系中分离所述烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12),少量的蒽醌杂质不影响所述烷基蒽醌组合物的性质。
189.根据本发明,如上所述,由原料蒽经烷基化反应制备蒽烷基化产物,分离其中的烷基化催化剂后得到反应液,其中的烷基蒽物系c
14+nh10+2n
(2≤n≤20)经氧化,会生产多种烷基蒽醌的混合物,即得到烷基蒽醌物系c
14+nh8+2n
o2(2≤n≤20)。所述烷基蒽醌物系中含有本
发明所需要的烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12),因此,需要进一步通过减压蒸馏的方法,从烷基蒽醌物系中分离出所述烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)。
190.根据本发明的方式3中,如图7所示,烷基蒽醌组合物的分离步骤中,通过蒸馏从烷基蒽醌物系中分离出所述烷基蒽醌组合物c
14+nh8+2n
o2,7≤n≤12的方法包括:
191.方式3a:
192.在烷基蒽醌物系中,沸点小于c
21h22
o2的沸点的物质的含量小于或等于1重量%;优选地,沸点小于c
21h22
o2的沸点的物质的含量小于或等于5重量%;从烷基蒽醌物系中蒸馏分离出烷基蒽醌组合物c
14+nh8+2n
o2,7≤n≤12的蒸馏条件包括:蒸馏塔顶压力为0.005-20kpa,塔底温度为230-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为240-430℃,理论板数为30-75,塔顶回流比为1-7;
193.方式3b:
194.在烷基蒽醌物系中,沸点小于c
21h22
o2的沸点的物质的含量高于或等于10重量%,优选地,沸点小于c
21h22
o2的沸点的物质的含量高于5重量%;先从从烷基蒽醌物系中蒸馏分离出沸点低于c
21h22
o2的物质,蒸馏条件包括:蒸馏塔顶压力为0.01-20kpa,塔底温度为230-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为240-430℃,理论板数为30-75,塔顶回流比为1-7;从除去沸点低于c
21h22
o2的物质的烷基蒽醌物系中蒸馏分离出烷基蒽醌组合物c
14+nh8+2n
o2,7≤n≤12的蒸馏条件包括:蒸馏塔顶压力为0.005-20kpa,塔底温度为230-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为240-430℃,理论板数为30-75,塔顶回流比为1-7。
195.在烷基蒽醌物系中,沸点小于c
21h22
o2的沸点的物质的含量大于1重量%且小于10重量%,采用方式3a或方式3b中的任意一种分离方式,优选地,沸点小于c
21h22
o2的沸点的物质的含量小于或等于5重量%,采用方式3a的分离方式;沸点小于c
21h22
o2的沸点的物质的含量高于5重量%,采用方式3b的分离方式。
196.根据本发明,根据方式4,若在原料蒽经烷基化反应进行蒽烷基化反应过程中,蒽没有完全被转化,烷基蒽氧化产物中沸点大于等于蒽醌的混合物内含蒽醌,其中蒽醌的含量高于或等于10重量%,优选地,蒽醌的含量高于5重量%,在分离轻组分之后,则需要先将蒽醌分离除去,然后再分离所述烷基蒽醌组合物。
197.根据物性分析可知,蒽醌的沸点为377℃,烷基蒽醌产物与蒽醌属同系物,彼此间存在沸点差异,可通过减压蒸馏技术来实现产物分离。但技术难点在于,蒽醌的熔点高达286℃,单独采用减压蒸馏技术来分离高熔点的蒽醌,操作难度大,管路极易发生堵塞问题,严重影响工艺的连续稳定运行。另外,蒽醌极易升华,升华过程难以控制,管路发生堵塞的机会显著增加。因此,单纯采用减压蒸馏技术来实现蒽醌-烷基蒽醌产物的分离是不切实际的。
198.因此,类似蒽和烷基蒽分离的过程,本发明人提出溶剂辅助分离蒽醌和蒸馏分离烷基蒽醌物系的方法。烷基蒽醌由于侧链取代基团的存在,破坏了蒽醌环结构的规整性,使得烷基蒽醌产物熔点明显降低,降低了后续蒸馏分离的难度。为此,本发明的发明人提出先采用溶剂辅助蒸馏技术,将熔点最高且最难实现分离操作的蒽醌分离除去,而后针对高沸点的烷基蒽醌物系,根据沸点差异,采用减压蒸馏技术实现进一步分离。
199.根据本发明的一种具体实施方式,如图2和图4所示,蒸馏溶剂辅助分离蒽醌在蒸
馏塔内进行。具体来说,预分离后,将含有蒽醌和烷基蒽醌物系的混合物引入蒸馏塔,该蒸馏过程可以是间歇式的,也可以是连续式的。蒸馏时,向蒸馏塔引入蒸馏溶剂,蒽醌在蒸馏条件下开始逐渐蒸出,同时引入的蒸馏溶剂进入蒸馏塔后也开始大量气化,并且与蒽醌一同蒸出进入塔顶冷凝器内进行冷凝。在大量的气化和液化的蒸馏溶剂分子氛围下,蒽醌无法经凝华和凝固结晶,而是溶解在蒸馏溶剂中形成溶液并随之一起流动,进而解决了蒽醌易堵塞管路的问题。蒸馏溶剂与蒽醌形成的溶液部分回流进入蒸馏塔重复蒸馏,部分流入塔顶产品罐收集。通过蒸馏溶剂的引入,控制其在塔顶与塔顶冷凝器间循环,同时调控进料位置、温度和用量,使之溶解蒽形成溶液一同顺利采出,即可实现蒽醌的高效分离,又可解决蒽醌蒸馏时的高度易凝的难题。
200.因此,根据本发明,在蒸馏溶剂辅助分离蒽醌过程中,所述蒸馏溶剂为在辅助分离蒽醌的过程中能够溶解蒽醌的、沸点介于100-340℃的有机溶剂。
201.优选地,所述蒸馏溶剂为沸点介于200-340℃的有机溶剂,可选自c
12-c
19
的直链烷烃和/或支链烷烃、卤代烃、芳香烃、醇、酮、酯和醚中的一种或多种;更优选地,所述烷烃为c
12-c
17
的直链烷烃和/或支链烷烃中的一种或多种;更优选地,所述卤代烃选自三氯苯、四氯苯、三溴苯、四溴苯、氯代c
10-c
18
烷、溴代c
10-c
18
烷中的一种或多种;更优选地,所述芳香烃为苯的烷基取代物,取代烷基的总碳数为4-12;进一步优选为丁基苯、戊基苯、己基苯、庚基苯、辛基苯、壬基苯、癸基苯、十一烷基苯、十二烷基苯、三乙基苯、四乙基苯、二丙基苯、三丙基苯、二丁基苯、二戊基苯中的一种或多种;更优选地,所述芳烃烷为苯的取代物,进一步优选为二苯甲烷及其烷基取代物,二苯乙烷及其烷基取代物;更优选为二苯甲烷、甲基二苯甲烷、1,2-二苯乙烷的一种或多种;更优选地,所述芳烃烷为萘或萘的烷基取代物,萘的取代烷基总碳数为1-4;进一步优选萘、甲基萘、二甲基萘、乙基萘、二乙基萘、丙基萘、甲基乙基萘、丁基萘中的一种或多种;更优选地,所述醇选自苯甲醇、丙三醇、二甘醇、三甘醇、四甘醇中的一种或多种;更优选地,所述酮选自1,1,3-三甲基环己烯酮、n-甲基吡咯烷酮、1,3-二甲基-2-咪唑啉酮中的一种或多种;更优选地,所述酯选自二甲酸酯、苯甲酸乙酯、邻苯二甲酸二甲酯、邻苯二甲酸二丁酯、乙二醇碳酸酯、丙二醇碳酸酯、磷酸三辛酯中的一种或多种;更优选地,所述醚选自乙二醇单苯醚、二乙二醇单丁醚、二苯醚和环丁砜中的一种或多种。
202.根据本发明,方式4中,蒸馏溶剂辅助分离蒽醌的条件包括:蒸馏塔顶压力为0.5-40kpa,塔底温度为230-450℃,理论板数为12-55,塔顶回流比为0.1-4;优选地,蒸馏塔顶压力为1-20kpa,塔底温度为260-430℃,理论板数16-50,塔顶回流比为0.2-1。所述蒸馏溶剂的用量可以根据进行蒸馏的含有蒽醌和烷基蒽醌物系的混合物中蒽醌的含量进行选择,以能够实现充分分离蒽醌以提高烷基蒽醌物系纯度为准。优选地,蒸馏溶剂与蒽醌的质量比为0.1:1-30:1。在确保能够获得令人满意的烷基蒽醌物系的纯度的条件下,从进一步降低本发明的方法的成本的角度出发,蒸馏溶剂与蒽醌的质量比为1:1-15:1。
203.根据本发明,在蒸馏溶剂辅助分离蒽醌过程中,塔顶收集的产品为蒸馏溶剂和蒽醌的混合物,需要将两者全部或部分的分离。优选情况下,所述蒸馏溶剂辅助分离蒽醌的步骤中还可以包括:收集含有蒽醌和蒸馏溶剂的混合物,并将蒽醌和蒸馏溶剂分离,回收得到蒽醌,并可以将蒸馏溶剂重复再利用。从蒸馏溶剂和蒽醌的混合物中分离蒽醌和蒸馏溶剂可以依据溶解度的差异,采用包括萃取和结晶的方法;也可以依据沸点的差异,采用蒸馏的方法。
204.根据本发明,优选采用蒸馏的方法分离蒸馏溶剂和蒽醌。所述蒸馏可以采用本领域公知的各种蒸馏设备,例如:筛板塔或者填料塔,更优选填料塔。具体来说,将含有蒽醌和蒸馏溶剂的混合物进行蒸馏,蒸馏条件包括:塔顶压力为1-100kpa,塔底温度为160-350℃,理论板数为6-40,塔顶回流比为0.1-3;进一步优选,塔顶压力为20-60kpa,塔底温度为200-310℃,理论板数为8-30,塔顶回流比为0.2-2。
205.根据本发明,如上所述,由原料蒽经烷基化反应制备蒽烷基化产物,分离其中的烷基化催化剂后和分离或不分离烷基化反应溶剂后得到的混合物,其中的烷基蒽物系c
14+nh10+2n
(2≤n≤20)经氧化,会生产多种烷基蒽醌的混合物,即得到烷基蒽醌物系c
14+nh8+2n
o2(2≤n≤20)。所述烷基蒽醌物系中含有本发明所需要的烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12),因此,需要进一步通过减压蒸馏的方法,从烷基蒽醌物系中分离出所述烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)。
206.根据本发明的方式4中,如图8所示,通过蒸馏从烷基蒽醌物系中分离出烷基蒽醌组合物c
14+nh8+2n
o2,7≤n≤12的方法包括:
207.方式4a:
208.在烷基蒽醌物系中,沸点小于c
21h22
o2的沸点的物质的含量小于或等于1重量%;优选地,沸点小于c
21h22
o2的沸点的物质的含量小于或等于5重量%;从烷基蒽醌物系中蒸馏分离出烷基蒽组合物c
14+nh8+2n
o2,7≤n≤12的蒸馏条件包括:蒸馏塔顶压力为0.005-20kpa,塔底温度为230-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选地,塔顶压力为0.05-10kpa,塔底温度为240-430℃,理论板数为30-75,塔顶回流比为1-7;
209.方式4b:
210.在烷基蒽醌物系中,沸点小于c
21h22
o2的沸点的物质的含量高于或等于10重量%,优选地,沸点小于c
21h22
o2的沸点的物质的含量高于5重量%;先从烷基蒽醌物系中蒸馏分离出沸点低于c
21h22
o2物质,蒸馏条件包括:蒸馏塔顶压力为0.01-20kpa,塔底温度为230-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选的,塔顶压力为0.05-10kpa,塔底温度为240-430℃,理论板数为30-75,塔顶回流比为1-7;从除去沸点低于c
21h22
o2物质的烷基蒽醌物系中蒸馏分离烷基蒽醌组合物c
14+nh8+2n
o2,7≤n≤12的蒸馏条件包括:蒸馏塔顶压力为0.005-20kpa,塔底温度为230-450℃,理论板数为20-90,塔顶回流比为0.5-8;优选的,塔顶压力为0.05-10kpa,塔底温度为240-430℃,理论板数为30-75,塔顶回流比为1-7。
211.在烷基蒽醌物系中,沸点小于c
21h22
o2的沸点的物质的含量大于1重量%且小于10重量%,采用方式4a或方式4b中的任意一种分离方式,优选地,沸点小于c
21h22
o2的沸点的物质的含量小于或等于5重量%,采用方式4a的分离方式;沸点小于c
21h22
o2的沸点的物质的含量高于5重量%,采用方式4b的分离方式。
212.根据本发明,在烷基蒽氧化反应过程中,反应的不同方法和操作条件,可能会带入或产生沸点低于蒽醌的其他物质以及氧化催化剂。其中,所述沸点低于蒽醌的物质含有氧化反应溶剂和氧化剂以及氧化反应副产物,统称为轻组分。因此,在方式3的烷基蒽醌组合物的分离之前,或者在方式4的在分离蒽醌和烷基蒽醌组合物之前,还包括分离轻组分的步骤即预分离。
213.根据本发明,所述分离轻组分的方法可以采用本领域常规的分离方法。优选情况下,从进一步提高分离效率以及操作简便的角度考虑,采用常压或减压蒸馏的方法分离含
有沸点低于蒽醌的轻组分、选择性含有的蒽醌和烷基蒽醌物系的混合物中的轻组分。
214.根据本发明的一种具体实施方式,方式3或方式4中,预分离的方法包括:将含有沸点低于蒽醌的物质、选择性含有的蒽醌和含有烷基蒽醌的物系的混合物进行蒸馏,得到含有沸点低于蒽醌的物质的馏出物以及含有选择性含有的蒽醌和烷基蒽醌物系的塔底产物,蒸馏的条件包括:蒸馏温度为50-390℃,优选为60-340℃,蒸馏的压力为0.1-20kpa,优选为0.5-15kpa。此外,可以将分离得到的轻组分按照反应的要求循环使用或者收集处理。
215.根据本发明,由于所述烷基蒽氧化产物中还含有氧化催化剂,因此,为了保证后续步骤的分离效果,优选情况下,所述方法还包括在方式3或方式4的预分离之前,先分离氧化催化剂。所述分离氧化催化剂的方法可以采用本领域常规的分离方法,例如沉降、过滤和离心分离中的一种或多种。
216.根据本发明,获得的烷基蒽醌组合物c
14+nh8+2n
o2(7≤n≤12)为目标产物,如其中仍含有其他杂质,可进一步通过其他常规分离方法或组合分离方法进行提纯,包括蒸馏、萃取和结晶。
217.以下将通过实施例对本发明进行详细描述。
218.物质组成数据采用色谱分析的方法获得。
219.(一)在蒽的烷基化反应中,采用各物质的色谱峰面积百分比来表示该物质的质量分数x,再结合摩尔质量,计算各物质的基于摩尔量的分数w(摩尔%)。用an表示蒽,ci-an表示烷基总碳数为i的烷基蒽。
220.蒽转化率x1(摩尔%)计算如式1所示:
[0221][0222]
烷基蒽组合物c
14+nh10+2n
(7≤n≤12)中,分子式为c
21h24
的物质统一记为a1、分子式为c
22h26
的物质统一记为a2、分子式为c
23h28
的物质统一记为a3、分子式为c
24h30
的物质统一记为a4、分子式为c
25h32
的物质统一记为a5、分子式为c
26h34
的物质统一记为a6。
[0223]
组合物中ai的质量分数gi(重量%)的计算式如式2所示:
[0224][0225]
烷基蒽组合物c
14+nh10+2n
(7≤n≤12)选择性s(摩尔%)的计算式如式3所示:
[0226][0227]
(二)在蒽-烷基蒽混合物分离过程中,某物质的纯度b为该物质的质量分数,分离出的蒽的纯度为b1,分离出的烷基蒽组合物的纯度为b2,均基于色谱分析数据得出。蒽的分离收率定义为y1,某烷基蒽组合物的分离收率定义为y2。
[0228]
(三)在烷基蒽的氧化反应过程中,定义ci-an转化率为x2(摩尔%),基于摩尔量计算的物质选择性为s(摩尔%)。采用各物质的色谱峰面积百分比来表示其质量分数,再结合摩尔质量,计算各物质的基于摩尔量的分数w,(摩尔%)。
[0229]
采用ci-an表示烷基蒽、ci-ao表示烷基蒽醌、ci-x表示其他副产物。
[0230]
烷基蒽组合物的转化率如式4所示:
[0231][0232]
烷基蒽醌组合物的选择性如式5所示:
[0233][0234]
烷基蒽醌组合物的氧化反应收率如式6所示:
[0235]yci-ao
=x2×sci-ao
ꢀꢀ
(6)
[0236]
烷基蒽醌组合物中各物质的含量计算,同烷基蒽组合物中各物质含量计算方法相同。
[0237]
实施例1-16用于说明本发明提供的烷基蒽组合物及其制备方法、分离方法。对比例1与实施例7进行对比说明本发明提供的分离方法特性。
[0238]
实施例1
[0239]
(一)烷基化反应
[0240]
蒽与乙烯烷基化反应,均三甲苯为溶剂,甲磺酸为催化剂。在搅拌釜中加入蒽173g、均三甲苯800ml、甲磺酸55g。密封后,在转速为1000转/分下升温至120℃,压力为0.5mpa。温度达到要求后,向釜内加入烯烃160g,进料时长为6h。当烯烃进料完毕后,维持反应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0241]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
22h26
的物质结构为蒽环与2个叔丁基相连(38重量%)、蒽环与1个乙基和1个己基相连(22重量%)、蒽环与1个叔丁基和2个乙基相连(40重量%);c
23h28
的物质结构为蒽环与1个叔丁基和1个叔戊基相连(52重量%)、蒽环与1个叔戊基和2个乙基相连(48重量%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(23重量%)、蒽环与2个叔戊基相连(8重量%)、蒽环与1个己基和2个乙基相连(30重量%)、蒽环与1个乙基和2个叔丁基相连(39重量%);c
25h32
的物质结构为蒽环与1个叔戊基和1个己基相连(100重量%);c
26h34
的物质结构为蒽环与2个己基相连(26重量%)、蒽环与1个乙基、1个叔丁基和1个己基相连(37重量%)、蒽环与3个叔丁基相连(47重量%)。
[0242]
(二)分离
[0243]
在压力为3kpa(绝对压力)、温度为60-150℃条件下,将沸点低于蒽的物质通过蒸馏去除后(下同),将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为262℃、理论板数为40、塔顶回流比为0.25、蒸馏溶剂与蒽的质量比为3:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为269℃、理论板数为65、塔顶回流比为3,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为289℃、理论板数为65、塔顶回
流比为3,塔顶收集产品,塔底收集其余重组分。
[0244]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0245]
实施例2
[0246]
(一)烷基化反应
[0247]
蒽与丙烯烷基化反应,均三甲苯为溶剂,甲磺酸为催化剂。在搅拌釜中加入蒽173g、均三甲苯800ml、甲磺酸55g。密封后,在转速为1000转/分下升温至120℃,压力为0.5mpa。温度达到要求后,向釜内加入烯烃163g,进料时长为6h。当烯烃进料完毕后,维持反应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0248]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
21h24
的物质结构为蒽环与1个庚基相连(100重量%);c
22h26
的物质结构为蒽环与1个丙基和1个叔戊基相连(86重量%)、蒽环与2个叔丁基相连(14重量%);c
23h28
的物质结构为蒽环与1个丙基和1个己基相连(45重量%)、蒽环与1个叔丁基和1个叔戊基相连(11重量%);蒽环与三个丙基相连(44%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(51重量%)、蒽环与2个叔戊基相连(4重量%)、蒽环与2个丙基和1个叔丁基相连(45重量%);c
25h32
的物质结构为蒽环与1个叔戊基和1个己基相连(25重量%)、蒽环与1个丙基和2个叔丁基相连(53重量%)、蒽环与1个叔戊基和2个丙基相连(22重量%);c
26h34
的物质结构为蒽环与2个己基相连(49重量%)、蒽环与1个己基和2个丙基相连(51重量%)。
[0249]
(二)分离
[0250]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为267℃、理论板数为40、塔顶回流比为0.25、蒸馏溶剂与蒽的质量比为3:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为273℃、理论板数为65、塔顶回流比为3,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为298℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0251]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0252]
实施例3
[0253]
(一)烷基化反应
[0254]
蒽与异丁烯烷基化反应,均三甲苯为溶剂,甲磺酸为催化剂。在搅拌釜中加入蒽173g、均三甲苯800ml、甲磺酸55g。密封后,在转速为1000转/分下升温至120℃,压力为0.5mpa。温度达到要求后,向釜内加入烯烃181g,进料时长为6h。当烯烃进料完毕后,维持反
应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0255]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
21h24
的物质结构为蒽环与1个庚基相连(100重量%);c
22h26
的物质结构为蒽环与2个叔丁基相连(98重量%)、蒽环与1个辛基相连(2重量%);c
23h28
的物质结构为蒽环与1个叔丁基和1个叔戊基相连(100重量%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(70重量%)、蒽环与2个叔戊基相连(30重量%);c
25h32
的物质结构为蒽环与1个叔戊基和1个己基相连(100重量%);c
26h34
的物质结构蒽环与2个己基相连(18重量%)、蒽环与3个叔丁基相连(82重量%)。
[0256]
(二)分离
[0257]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为269℃、理论板数为40、塔顶回流比为0.25、蒸馏溶剂与蒽的质量比为3:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为303℃、理论板数为65、塔顶回流比为3,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为297℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0258]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0259]
实施例4
[0260]
(一)烷基化反应
[0261]
蒽与2-甲基-2-丁烯烷基化反应,均三甲苯为溶剂,甲磺酸为催化剂。在搅拌釜中加入蒽173g、均三甲苯800ml、甲磺酸27g。密封后,在转速为1000转/分下升温至120℃,压力为0.2mpa。温度达到要求后,向釜内加入烯烃97g,进料时长为6h。当烯烃进料完毕后,维持反应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0262]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
21h24
的物质结构为蒽环与1个庚基相连(100重量%);c
22h26
的物质结构为蒽环与2个叔丁基相连(100重量%);c
23h28
的物质结构为蒽环与1个叔丁基和1个戊基相连(98重量%)、蒽环与1个壬基相连(2重量%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(12重量%)、蒽环与2个戊基相连(85重量%),蒽环与1个癸基相连(3重量%);c
25h32
的物质结构为蒽环与1个戊基和1个己基相连(100重量%);c
26h34
的物质结构蒽环与2个己基相连(36重量%)、蒽环与3个叔丁基相连(64重量%)。
[0263]
(二)分离
[0264]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为270℃、理论板数为40、塔顶回流比为0.25、蒸馏溶剂与蒽的质
量比为3:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为292℃、理论板数为65、塔顶回流比为3,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为298℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0265]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0266]
实施例5
[0267]
(一)烷基化反应
[0268]
蒽与2-甲基-2-戊烯烷基化反应,均三甲苯为溶剂,甲磺酸为催化剂。在搅拌釜中加入蒽173g、均三甲苯800ml、甲磺酸27g。密封后,在转速为1000转/分下升温至120℃,压力为0.2mpa。温度达到要求后,向釜内加入烯烃408g,进料时长为6h。当烯烃进料完毕后,维持反应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0269]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
21h24
的物质结构为蒽环与1个庚基相连(100重量%);c
22h26
的物质结构为蒽环与2个叔丁基相连(100重量%);c
23h28
的物质结构为蒽环与1个叔丁基和1个叔戊基相连(100重量%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(68重量%)、蒽环与2个叔戊基相连(32重量%);c
25h32
的物质结构为蒽环与1个叔戊基和1个己基相连(66重量%)、蒽环与1个叔丁基和1个庚基相连(34重量%);c
26h34
的物质结构蒽环与2个己基相连(75重量%)、蒽环与1个叔戊基和1个庚基相连(10重量%)、蒽环与3个叔丁基相连(15重量%)。
[0270]
(二)分离
[0271]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为285℃、理论板数为40、塔顶回流比为0.25、蒸馏溶剂与蒽的质量比为3:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为315℃、理论板数为65、塔顶回流比为3,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为308℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0272]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0273]
实施例6
[0274]
(一)烷基化反应
[0275]
蒽与2-甲基-2-丁烯烷基化反应,均三甲苯为溶剂,对甲基苯磺酸为催化剂。在搅
拌釜中加入蒽77g、均三甲苯800ml、对甲基苯磺酸8g。密封后,在转速为1000转/分下升温至100℃,压力为0mpa。温度达到要求后,向釜内加入烯烃30g,进料时长为6h。当烯烃进料完毕后,维持反应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0276]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
21h24
的物质结构为蒽环与1个庚基相连(100重量%);c
22h26
的物质结构为蒽环与2个叔丁基相连(100重量%);c
23h28
的物质结构为蒽环与1个叔丁基和1个戊基相连(99重量%)、蒽环与1个壬基相连(1重量%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(9重量%)、蒽环与2个戊基相连(90重量%),蒽环与1个癸基相连(1重量%);c
25h32
的物质结构为蒽环与1个戊基和1个己基相连(100重量%);c
26h34
的物质结构蒽环与2个己基相连(30重量%)、蒽环与3个叔丁基相连(70重量%)。
[0277]
(二)分离
[0278]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为240℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为284℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0279]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0280]
实施例7
[0281]
(一)烷基化反应
[0282]
蒽与2-甲基-2-丁烯烷基化反应,均三甲苯为溶剂,对甲基苯磺酸为催化剂。在搅拌釜中加入蒽297g、均三甲苯800ml、对甲基苯磺酸174g。密封后,在转速为1000转/分下升温至140℃,压力为0.5mpa。温度达到要求后,向釜内加入烯烃292g,进料时长为6h。当烯烃进料完毕后,维持反应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0283]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
21h24
的物质结构为蒽环与1个庚基相连(100重量%);c
22h26
的物质结构为蒽环与2个叔丁基相连(100重量%);c
23h28
的物质结构为蒽环与1个叔丁基和1个戊基相连(96重量%)、蒽环与1个壬基相连(4重量%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(17重量%)、蒽环与2个戊基相连(80重量%),蒽环与1个癸基相连(3重量%);c
25h32
的物质结构为蒽环与1个戊基和1个己基相连(100重量%);c
26h34
的物质结构蒽环与2个己基相连(37重量%)、蒽环与3个叔丁基相连(63重量%)。
[0284]
(二)分离
[0285]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0286]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0287]
对比例1
[0288]
与实施例7对比,步骤(一)烷基化反应同实施例7相同。
[0289]
(二)分离
[0290]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)直接蒸馏分离蒽。蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3。塔顶收集蒽,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0291]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0292]
实施例8
[0293]
(一)烷基化反应
[0294]
蒽与2-甲基-2-丁烯烷基化反应,均三甲苯为溶剂,对甲基苯磺酸为催化剂。在搅拌釜中加入蒽460g、均三甲苯800ml、对甲基苯磺酸493g。密封后,在转速为1000转/分下升温至165℃,压力为1.0mpa。温度达到要求后,向釜内加入烯烃452g,进料时长为6h。当烯烃进料完毕后,维持反应条件不变继续反应6h,而后终止反应。同样条件反应多批次,沉降分离催化剂后,统一收集反应产物作为烷基蒽分离的原料。
[0295]
基于反应机理和质谱结果,分析烷基蒽产物中分子式为c
21h24
的物质结构为蒽环与1个庚基相连(100重量%);c
22h26
的物质结构为蒽环与2个叔丁基相连(100重量%);c
23h28
的物质结构为蒽环与1个叔丁基和1个戊基相连(98重量%)、蒽环与1个壬基相连(2重量%);c
24h30
的物质结构为蒽环与1个叔丁基和1个己基相连(10重量%)、蒽环与2个戊基相连(87重量%),蒽环与1个癸基相连(3重量%);c
25h32
的物质结构为蒽环与1个戊基和1个己基相连(100重量%);c
26h34
的物质结构蒽环与2个己基相连(33重量%)、蒽环与3个叔丁基
相连(67重量%)。
[0296]
(二)分离
[0297]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为258℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为316℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为304℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0298]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表1所示。
[0299]
实施例9
[0300]
步骤(一)烷基化反应同实施例7相同。
[0301]
(二)分离
[0302]
将沸点低于蒽的物质通过常压蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,4-三氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为12:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0303]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0304]
实施例10
[0305]
步骤(一)烷基化反应同实施例7相同。
[0306]
(二)分离
[0307]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为二苯甲烷,蒸馏条件:塔顶压力为3kpa、塔底温度为259℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为15:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理
论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0308]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0309]
实施例11
[0310]
步骤(一)烷基化反应同实施例7相同。
[0311]
(二)分离
[0312]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为2,7-二甲基萘,蒸馏条件:塔顶压力为8kpa、塔底温度为328℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为3:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0313]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0314]
实施例12
[0315]
步骤(一)烷基化反应同实施例7相同。
[0316]
(二)分离
[0317]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为305℃、理论板数为65、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0318]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0319]
实施例13
[0320]
步骤(一)烷基化反应同实施例7相同。
[0321]
(二)分离
[0322]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连
续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为3kpa、塔底温度为338℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0323]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0324]
实施例14
[0325]
步骤(一)烷基化反应同实施例7相同。
[0326]
(二)分离
[0327]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为40、塔顶回流比为3,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为301℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0328]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0329]
实施例15
[0330]
步骤(一)烷基化反应同实施例7相同。
[0331]
(二)分离
[0332]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.8kpa、塔底温度为297℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0333]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0334]
实施例16
[0335]
步骤(一)烷基化反应同实施例7相同。
[0336]
(二)分离
[0337]
将沸点低于蒽的物质通过蒸馏去除后,将蒽和烷基蒽的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽。蒸馏溶剂为1,2,3,4-四氯苯,蒸馏条件:塔顶压力为3kpa、塔底温度为276℃、理论板数为40、塔顶回流比为0.3、蒸馏溶剂与蒽的质量比为10:1。塔顶收集蒸馏溶剂与蒽的混合物,塔底收集的烷基蒽混合物继续蒸馏。2)蒸馏分离沸点低于c
21h24
的物质。蒸馏条件:塔顶压力为1kpa、塔底温度为310℃、理论板数为55、塔顶回流比为4,塔顶收集沸点低于低于c
21h24
的物质,塔底收集的混合物继续蒸馏。3)蒸馏分离产物烷基蒽组合物c
14+nh10+2n
(7≤n≤12)。蒸馏条件:塔顶压力为0.4kpa、塔底温度为291℃、理论板数为65、塔顶回流比为3,塔顶收集产品,塔底收集其余重组分。
[0338]
步骤(一)中蒽的转化率x1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的组成gi、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的选择性s。步骤(二)中分离得到的蒽的纯度b1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的纯度b2、蒽的分离收率y1、烷基蒽组合物c
14+nh10+2n
(7≤n≤12)的分离收率y2如表2所示。
[0339]
[0340][0341]
实施例17-32用于说明本发明提供的烷基蒽醌组合物及其制备方法,对比例2用于比较实施例20中的烷基蒽醌组合物与其他蒽醌制成工作液后的加氢性能。
[0342]
实施例17
[0343]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例1。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0344]
(三)氧化反应。
[0345]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、铬酸钾239g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为96.67摩尔%,组成见表3。氧化产物烷基蒽醌(蒽醌母核为9,10-蒽醌)的烷基取代基结构及取代位置与对应的烷基蒽的烷基取代基结构及取代位置相同,下同。
[0346]
实施例18
[0347]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例2。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0348]
(三)氧化反应。
[0349]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、六水硝酸镧213g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为96.81摩尔%,组成见表3。
[0350]
实施例19
[0351]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例3。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0352]
(三)氧化反应。
[0353]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、钼酸钠169g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为95.94摩尔%,组成见表3。
[0354]
实施例20
[0355]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0356]
(三)氧化反应。
[0357]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、叔丁醇2000g、二氧化锆101g。反应在常压80℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为96.58摩尔%,组成见表3。
[0358]
实施例21
[0359]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例5。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0360]
(三)氧化反应。
[0361]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、叔
丁醇2000g、铬酸钾239g。反应在常压80℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为96.41摩尔%,组成见表3。
[0362]
实施例22
[0363]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例6。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0364]
(三)氧化反应。
[0365]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、钼酸钠169g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为94.3摩尔%,组成见表3。
[0366]
实施例23
[0367]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例7。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0368]
(三)氧化反应。
[0369]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、钼酸钠169g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为95.51摩尔%,组成见表3。
[0370]
实施例24
[0371]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例8。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0372]
(三)氧化反应。
[0373]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、钼酸钠169g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为95.2摩尔%,组成见表3。
[0374]
表3
[0375][0376]
实施例25
[0377]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物
为原料氧化制备烷基蒽醌组合物。
[0378]
(三)氧化反应。
[0379]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、叔丁醇2000g、二氧化锆101g。反应在常压80℃下进行,通过蠕动泵向釜内加入双氧水1374g(过氧化氢含量为30重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为84.01摩尔%。
[0380]
实施例26
[0381]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0382]
(三)氧化反应。
[0383]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物500g、n-甲基吡咯烷酮2000g、钼酸钠663g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水3282g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为60.18摩尔%。
[0384]
实施例27
[0385]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0386]
(三)氧化反应。
[0387]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n,n-二甲基甲酰胺2000g、铬酸钾239g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为94.98摩尔%。
[0388]
实施例28
[0389]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0390]
(三)氧化反应。
[0391]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、铬酸钾277g。反应在常压65℃下进行,通过蠕动泵向釜内加入双氧水973g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为58.44摩尔%。
[0392]
实施例29
[0393]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0394]
(三)氧化反应。
[0395]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、六水硝酸镧106g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水417g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为43.21摩尔%。
[0396]
实施例30
[0397]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0398]
(三)氧化反应。
[0399]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、n-甲基吡咯烷酮2000g、钼酸钠169g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水836g(过氧化氢含量为50重量%),进料总时长为3h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为62.61摩尔%。
[0400]
实施例31
[0401]
步骤(一)同实施例4。反应结束后,分出催化剂和沸点小于蒽的物质,再将蒽与烷基蒽的混合物一起送入步骤(二)进行氧化反应。
[0402]
(二)氧化反应
[0403]
将步骤(一)获得的蒽和烷基蒽的混合物加入反应釜,并加入n-甲基吡咯烷酮3478g、钼酸钠400g。反应在常压100℃下进行,通过蠕动泵向釜内加入双氧水1979g(过氧化氢含量为50重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。反应结束,分出催化剂和沸点小于蒽醌的物质,得到蒽醌和烷基蒽醌的混合物,送去步骤(三)进行分离。蒽醌和烷基蒽醌的氧化反应总收率为95.6摩尔%。
[0404]
(三)分离过程
[0405]
分离蒽醌和烷基蒽醌的混合物,获得烷基蒽醌组合物。
[0406]
将蒽醌和烷基蒽醌的混合物送入蒸馏塔进行连续蒸馏,物料流量为10g/min。1)溶剂辅助分离蒽醌:蒸馏溶剂为2,7-二甲基萘,蒸馏条件:塔顶压力为3kpa、塔底温度为298℃、理论板数为40、塔顶回流比为0.25、蒸馏溶剂与蒽的质量比为3:1。2)沸点低于c
21h22
o2物质的分离,蒸馏条件:塔顶压力为1kpa、塔底温度为326℃、理论板数为65、塔顶回流比为3。3)蒸馏获得目标产物c
14+nh8+2n
o2(7≤n≤12)。蒸馏条件:塔顶压力为0.7kpa、塔底温度为310℃、理论板数为65、塔顶回流比为3。分离结果:烷基蒽醌组合物的纯度为93.81重量%,分离收率为89.95重量%。
[0407]
实施例32
[0408]
步骤(一)蒽烷基化反应与步骤(二)分离同实施例4。以分离获得的烷基蒽组合物为原料氧化制备烷基蒽醌组合物。
[0409]
(三)氧化反应。
[0410]
烷基蒽组合物氧化制备烷基蒽醌组合物。向反应釜内加入烷基蒽组合物127g、甲醇2000g、36重量%盐酸208g。反应在常压65℃下进行,通过蠕动泵向釜内加入双氧水232g(过氧化氢含量为30重量%),进料总时长为8h。进料结束后,维持条件不变继续反应2h。烷基蒽醌组合物的氧化反应收率为94.2摩尔%。
[0411]
对比例2
[0412]
30℃条件下,配制三种烷基蒽醌工作液,a、b和c。混合溶剂为均三甲苯和二异叔丁基甲醇,体积比为3:2。
[0413]
工作液a的蒽醌载体为实施例20中的烷基蒽醌组合物,其中c
21h22
o2占3.96重量%、c
22h24
o2占12.57重量%、c
23h26
o2占30.88重量%、c
24h28
o2中占35.72重量%、c
25h30
o2占12.37重量%、c
26h32
o2占4.5重量%。工作液a中烷基蒽醌的总含量为1.2mol/l。
[0414]
工作液b的蒽醌载体为复合烷基蒽醌,其中c
17h14
o2占1.89重量%、c
18h16
o2占25.14重量%、c
19h18
o2占61.02重量%、c
20h20
o2中占11.88重量%、c
14
h8o2占0.07重量%。工作液b中烷基蒽醌的总含量为0.78mol/l。
[0415]
工作液c的蒽醌载体为2-叔戊基蒽醌和2-仲戊基蒽醌,两者摩尔比为3:1。工作液c中戊基蒽醌的含量为0.65mol/l。
[0416]
分别采用工作液a、b和c进行间歇搅拌釜加氢,测量工作液的极限氢化效率(简称极限氢效)。工作液加入量为120ml、催化剂pd/al2o3(pd含量为1.8重量%)加入量为0.6g、反应温度为60℃、釜内氢气压力为0.3mpa。向釜内连续通入氢气与工作液反应,直至氢蒽醌晶体析出,停止氢气进料。隔绝空气的条件下,先分离出氢蒽醌晶体和催化剂,再对饱和的氢化溶液进行氧化和萃取,测量氢化效率。
[0417]
氢化效率检测可按常规方法进行。例如:向氢化液中加入适量纯水和磷酸,在50℃条件下通入纯氧进行氧化,当有机相颜色由黑色转变为亮黄色后,用纯水对有机相多次进行萃取。分出并收集水相,加入适量20重量%硫酸,采用0.03mol/l高锰酸钾进行滴定测量过氧化氢含量,计算氢化效率,结果见表4。
[0418]
表4
[0419][0420]
本发明提出了一种高碳数多烷基取代的蒽醌组合物,并提供了制备方法。可以通过蒽烷基化反应与分离工艺制备烷基蒽组合物,再经氧化制备烷基蒽醌组合物。
[0421]
通过表1和表2的结果可以看出,本发明提供的烷基蒽组合物,可通过控制烷基化反应和分离来制备特定结构和组成的烷基蒽组合物,所得的烷基蒽组合物可经氧化方法制备烷基蒽醌组合物,本发明为合成新型复合烷基蒽醌提供了新的技术路线。
[0422]
本发明提供的蒽和烷基蒽分离的方法中,通过引入特定蒸馏溶剂,匹配特殊蒸馏工艺,实现溶剂溶解蒽并携带蒽一同流动分离,彻底解决了蒽分离过程中的易堵难题,实现了蒽的高效分离,提高了蒽的收率;针对烷基蒽混合物系高沸易凝、高温生焦的问题,开发的减压蒸馏工艺,可实现烷基蒽组合物的有效分离,产品纯度和分离收率高,分离难度低。
[0423]
通过对比例2的结果可看出,本发明提供的高碳数多烷基取代的混合蒽醌同单一蒽醌相比,具有优异的溶解性能和加氢性能;同低碳数烷基蒽醌组合物相比,溶解性能和加氢性能得到进一步的提升。
[0424]
本发明提出的高碳数多烷基取代的蒽醌组合物,拓宽和丰富了过氧化氢生产中的蒽醌载体种类,为该产业带来了新的载体类型。
[0425]
本发明提出的烷基蒽醌组合物的制备方法,清洁高效,工艺灵活性强,可针对性调配工艺,满足不同混合烷基蒽醌产品的需求。
[0426]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技
术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1