一种低VOC低气味聚丙烯树脂及其制备方法和应用与流程
一种低voc低气味聚丙烯树脂及其制备方法和应用
技术领域
1.本发明属于聚烯烃领域,具体地,涉及一种低voc低气味聚丙烯树脂,一种低voc低气味聚丙烯树脂的制备方法,由该制备方法制得的聚丙烯树脂,以及所述聚丙烯树脂在制备纤维或无纺布中的应用。
背景技术:
2.聚丙烯材料因具有相对密度小、良好的力学性能及加工性能、较高的耐热性和耐化学腐蚀,非常适合于满足多种不同应用中的需求。近年来,聚丙烯已经成为非织造布中使用最广泛的聚合物之一,主要用于卫生和医用制品、建筑业和农业、地毯和纺织品等。
3.用于非织造布的聚丙烯需要具有较高的熔体流动指数和较窄的分子量分布指数。同时,随着人们健康意识的提高,聚丙烯及其非织造布制品的voc和气味都期待得到更加有效的控制和降低。
4.最常见的生产高熔体流动指数聚丙烯的方法是向聚丙烯树脂中添加有机过氧化物控制降解,从而提高树脂的流动性,又称为可控流变技术。该技术采用有机过氧化物将聚丙烯树脂中高分子链断裂,使树脂分子量分布变窄,流动性大大改善,而不明显影响材料的其他性能。但是传统的过氧化物法在调节分子量的过程中会产生部分断链小分子副产物,从而导致聚丙烯粒料voc含量的提高。
5.近年来,新发展起来的高熔体流动指数聚丙烯的制备方法是借助高氢调敏感性外给电子体在聚合过程中使用氢气调节聚丙烯分子量及其分布,并在聚合釜中直接合成树脂产品。如专利文献cn101993599a中使用异丁基三乙氧基硅烷等外给电子体、专利文献cn102532381b和cn102532380b中利用非对称外给电子体技术直接聚合得到的聚丙烯熔指可达50-300g/10min。然而,使用氢调敏感性好的外给电子体固然可以提高聚丙烯的流动性,但通常也会造成聚丙烯模量较低。这是因为现有的ziegler-natta催化剂很难同时满足高氢调敏感性和高立构规整度。另外,现有高氢调性能ziegler-natta催化剂和/或外给电子体所制备的聚丙烯还存在分子量分布较宽的问题,导致含有较多的低分子量部分,进而提高了聚丙烯粒料及其制品的voc。
6.为改善原有聚丙烯材料中voc的散发问题,常用的方法有化学反应、物理吸附以及物理-化学方法。专利文献cn101570612b通过添加一种无机光催化剂分解有机小分子来降低voc含量,但组分复杂,效果也有限。cn101255252b通过添加有机物驱除剂(如异丙醇/水)来降低材料的voc,但该方法存在相容性和持久性方面的问题。cn1727389a公开了使用细孔硅胶和分子筛作为吸附剂来降低材料的气味和voc,但高温下吸附平衡会向解吸附方向移动,致使聚丙烯材料存在voc再释放的隐患。cn102276921b公开了加入植物纤维并通过物理吸附和化学键共同作用来降低聚丙烯材料的voc含量,但该方法会影响材料的性能。
7.综上,氢调法得到的聚丙烯模量较低,难以满足应用需求,且较宽的分子量分布会导致voc含量的升高;而物理方法、化学方法或物理-化学方法来降低voc含量也具有各自的局限性。
8.近十年来,非织造布行业展现出较大的市场前景。而丙纶,即聚丙烯纤维非织造布则占据了50%以上的产能。而且,随着医疗、卫生、汽车内饰、家居用品、过滤材料等应用市场的扩大,对聚丙烯纺粘非织造布的需求还将进一步增加。生产聚丙烯非织造布需要聚合物树脂具有较窄的分子量分布、较大的熔体流动指数以及较高的等规度;同时为满足制品在环保和安全性方面的要求,产品应当不含塑化剂同时具有较低的voc含量。因此,利用不含邻苯二甲酸酯(塑化剂)的催化剂体系,通过过氧化物可控流变技术得到高熔体流动指数的低voc、低气味纺粘纤维用聚丙烯树脂是目前亟需解决的一个问题。
技术实现要素:
9.本发明的目的是提供一种低voc低气味聚丙烯树脂及其制备方法和应用,该聚丙烯树脂通过可控流变技术制得,并且同时具有低voc和低气味。
10.本发明的第一方面提供一种低voc低气味聚丙烯树脂,该聚丙烯树脂具有以下特征:voc组分含量低于50μg
·
c/g;气味4.0级以下;所述聚丙烯树脂的分子量分布≤4.0,优选≤3.5。
11.本发明的第二方面提供一种低voc低气味聚丙烯树脂的制备方法,包括以下步骤:
12.步骤一:在ziegler-natta催化剂存在下,使丙烯、或者丙烯与除丙烯外的c
2-c
12
烯烃的混合物进行连续聚合,得到聚丙烯粉料;
13.所述ziegler-natta催化剂含有:
14.(i)固体催化剂组分,所述固体催化剂组分是以卤化镁和醇类化合物为溶解体系、以复合给电子体为助析出剂,采用颗粒成型工艺和内给电子体复配得到的反应物;
15.(ii)有机铝化合物;以及
16.(iii)外给电子体;所述外给电子体选自通式为rnsi(or')
4-n
的硅烷类化合物中的至少两种;式中0<n≤3,r选自氢原子、卤素、c
1-c
20
的烷基、c
3-c
20
的环烷基、c
6-c
20
的芳基或c
1-c
20
的卤代烷基,r'选自c
1-c
20
的烷基、c
3-c
20
的环烷基、c
6-c
20
的芳基或c
1-c
20
的卤代烷基;优选地,r选自氢原子、卤素、c
1-c
12
的烷基、c
3-c
12
的环烷基、c
6-c
12
的芳基或c
1-c
12
的卤代烷基,r'选自c
1-c
12
的烷基、c
3-c
12
的环烷基、c
6-c
12
的芳基或c
1-c
12
的卤代烷基;
17.步骤二:将上述聚丙烯粉料与复合助剂、过氧化物和气味吸附剂混合造粒,得到所述聚丙烯树脂;
18.其中,以聚丙烯粉料的重量为基准,所述复合助剂的加入量为1500-4000ppm,所述过氧化物的加入量为500-1000ppm,所述气味吸附剂的加入量为1500-5000ppm。
19.本发明的第三方面提供由上述制备方法制得的聚丙烯树脂。
20.本发明的第四方面提供上述聚丙烯树脂在制备纤维或无纺布中的应用。
21.本发明的技术效果体现在:在不改动现有工艺装置的前提下,采用高立构规整度、窄分子量分布催化剂搭配复配的硅烷类外给电子体得到聚丙烯粉末;利用聚丙烯粉末、复合助剂、过氧化物和气味吸附剂制备得到的聚丙烯树脂具有较窄的分子量分布、低voc含量以及低气味。
22.本发明的其它特征和优点将在随后具体实施方式部分予以详细说明。
具体实施方式
23.以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
24.本发明提供一种低voc低气味聚丙烯树脂,该聚丙烯树脂具有以下特征:voc组分含量低于50μg
·
c/g;气味4.0级以下;所述聚丙烯树脂的分子量分布≤4.0,优选≤3.5。
25.根据本发明,除上述特征外,优选地,所述聚丙烯树脂还具有以下特征:拉伸屈服应力≥29mpa,优选拉伸屈服应力≥32mpa,更优选拉伸屈服应力≥35mpa;弯曲模量≥1100mpa,优选弯曲模量≥1250mpa,更优选弯曲模量≥1500mpa;结晶温度为100-125℃,优选为115-120℃;熔融温度为140-170℃,优选为162-167℃;230℃、2.16kg载荷下的熔融指数为20-80g/10min,优选熔融指数为30-70g/10min,更优选熔融指数为35-60g/10min。对于均聚聚丙烯树脂,所述聚丙烯树脂的等规指数优选≥96.5%。
26.本发明中,voc组分含量按照德国汽车工业联合会的标准vda277测试。气味按照德国汽车工业联合会的标准vda270测试。分子量分布通过gpc测定。熔融指数根据astm d1238,在230℃、2.16kg载荷下测定。等规指数采用庚烷抽提法测定。拉伸强度根据astm d638测量注塑样品测得。弯曲模量根据astm d790测量注塑样品测得。结晶温度和熔融温度采用示差扫描量热法(dsc)测试。
27.本发明还提供一种低voc低气味聚丙烯树脂的制备方法,包括以下步骤:
28.步骤一:在ziegler-natta催化剂存在下,使丙烯、或者丙烯与除丙烯外的c
2-c
12
烯烃的混合物进行连续聚合,得到聚丙烯粉料;
29.所述ziegler-natta催化剂含有:
30.(i)固体催化剂组分,所述固体催化剂组分是以卤化镁和醇类化合物为溶解体系、以复合给电子体为助析出剂,采用颗粒成型工艺和内给电子体复配技术得到的反应物;
31.(ii)有机铝化合物;以及
32.(iii)外给电子体;所述外给电子体选自通式为rnsi(or')
4-n
的硅烷类化合物中的至少两种;式中0<n≤3,r选自氢原子、卤素、c
1-c
20
的烷基、c
3-c
20
的环烷基、c
6-c
20
的芳基或c
1-c
20
的卤代烷基,r'选自c
1-c
20
的烷基、c
3-c
20
的环烷基、c
6-c
20
的芳基或c
1-c
20
的卤代烷基;优选地,r选自氢原子、卤素、c
1-c
12
的烷基、c
3-c
12
的环烷基、c
6-c
12
的芳基或c
1-c
12
的卤代烷基,r'选自c
1-c
12
的烷基、c
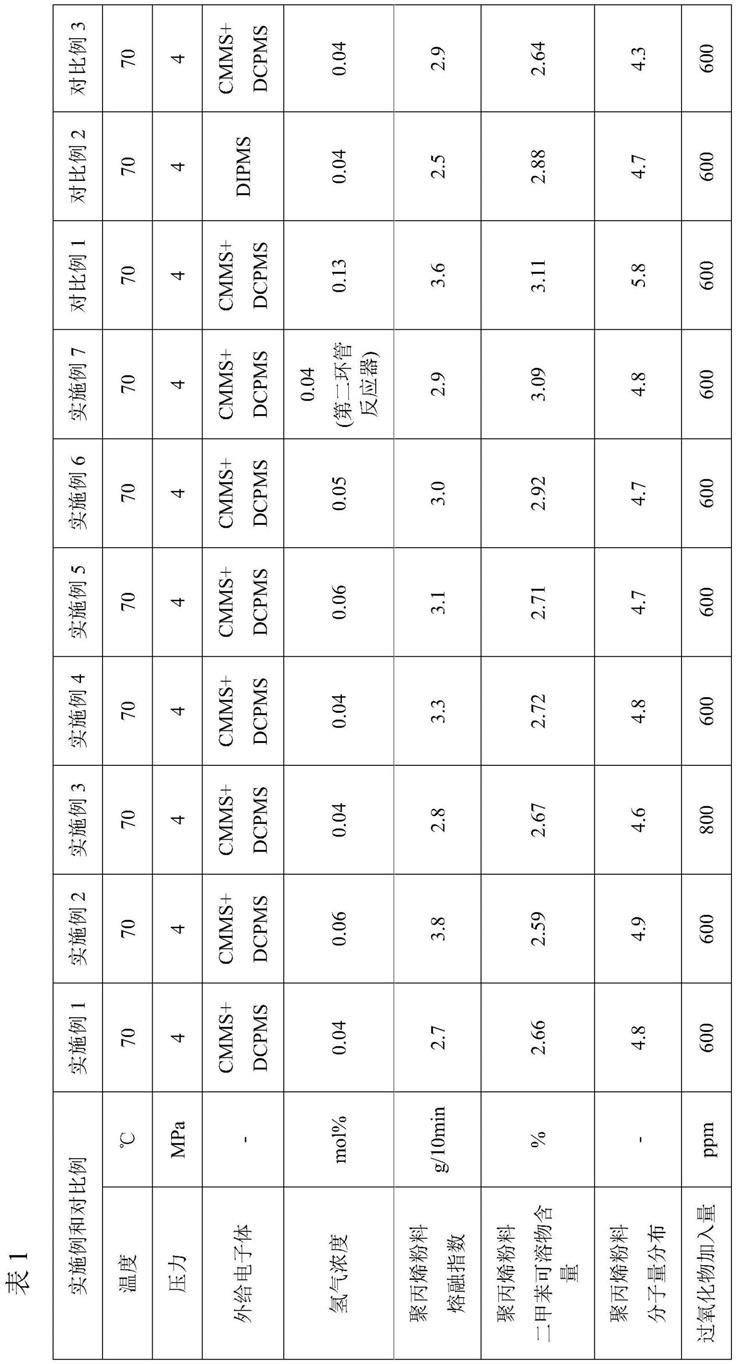
3-c
12
的环烷基、c
6-c
12
的芳基或c
1-c
12
的卤代烷基;
33.步骤二:将上述聚丙烯粉料与复合助剂、过氧化物和气味吸附剂混合造粒,得到所述聚丙烯树脂;
34.其中,以聚丙烯粉料的重量为基准,所述复合助剂的加入量为1500-4000ppm,所述过氧化物的加入量为500-1000ppm,所述气味吸附剂的加入量为1500-5000ppm。
35.根据本发明一种优选实施方式,所述复合助剂包括抗氧剂、吸酸剂和滑石粉,质量比为:
36.抗氧剂:吸酸剂:滑石粉=20:3:2-20:12:5。
37.所述抗氧剂优选为受阻酚类抗氧剂、亚磷酸酯类抗氧剂和季戊四醇酯类抗氧剂中的一种或多种;更优选地,所述受阻酚类抗氧剂选自四(β-(3,5-二叔丁基-4-羟基苯基)丙酸)季戊四醇酯)、β-(3,5-二叔丁基-4-羟基苯基)丙酸正十八碳醇酯、双(3,5-二叔丁基-4-羟基苄基膦酸单乙酯)钙、n,n'-双-(3-(3,5-二叔丁基-4-羟基苯基)丙酰基)己二胺、1,3,
5-三(3,5-二叔丁基-4-羟基苄基)异氰尿酸,所述亚磷酸酯类抗氧剂为(2,4-二叔丁基苯基)亚磷酸三酯、双(2,4-二叔丁基苯酚)季戊四醇二亚磷酸酯);进一步优选地,所述抗氧剂为受阻酚类抗氧剂和亚磷酸酯类抗氧剂的混合物,更进一步优选地,所述抗氧剂为双(3,5-二叔丁基-4-羟基苄基膦酸单乙酯)钙和(2,4-二叔丁基苯基)亚磷酸三酯的混合物;更优选地,双(3,5-二叔丁基-4-羟基苄基膦酸单乙酯)钙和(2,4-二叔丁基苯基)亚磷酸三酯的质量比为1:0.8-1.2。
38.本发明中,所述吸酸剂可商购获得,优选为硬脂酸盐类,进一步优选选自硬脂酸钙、硬脂酸锌和硬脂酸钠中的至少一种,最优选为硬脂酸钙。
39.本发明中,所述滑石粉的细度优选为2000-5000目。
40.本发明中,所述过氧化物可选自过氧化二叔丁基、过氧化二叔戊基、2,5-二甲基-2,5-双(叔丁基过氧基)己烷、2,5-二甲基-2,5双(过氧化氢)己烷、双(叔丁基过氧化异丙基)苯中的一种或两种的混合物;优选为过氧化二叔丁基和双(叔丁基过氧化异丙基)苯的混合物;更优选为过氧化二叔丁基和双(叔丁基过氧化异丙基)苯按8-12:1质量比混合的混合物。
41.本发明中,优选地,所述气味吸附剂为具有微孔的硅酸盐,所述硅酸盐优选为硅酸钠盐、硅酸铝盐、硅酸镁盐和硅酸钙盐中的至少一种,更优选为具有微孔的硅酸铝盐。所述具有微孔的硅酸盐的比表面积为200-700m2/g,优选比表面积为300-550m2/g;微孔的平均孔径为0.1-3nm,优选微孔的平均孔径为0.3-2nm。上述气味吸附剂可商购获得,例如商购自宁波嘉和公司的气味吸附剂jh100a,其为具有微孔结构的硅酸铝盐,比表面积为500m2/g,平均孔径为0.4nm。
42.根据本发明的方法,优选地,步骤一所得聚丙烯粉料的熔融指数为2.0-6.0g/10min,二甲苯可溶物含量≤3.0,分子量分布指数为4.5-5.2。
43.根据本发明一种优选实施方式,所述固体催化剂组分的制备步骤包括:(1)将卤化镁和醇类化合物在烃类溶剂的存在下进行第一接触反应,形成均匀溶液;(2)在助析出剂存在下,将步骤(1)所得的均匀溶液与第一部分钛化合物进行第二接触反应,得到含固体沉淀的混合物;(3)将步骤(2)所得的混合物与第一部分内给电子体化合物d进行第三接触反应,得到悬浮液;(4)将步骤(3)得到的悬浮液进行固液分离得到的固体组分与第二部分钛化合物、第二部分内给电子体化合物d进行第四接触反应,再过滤出液体,得到固体产物;(5)将步骤(4)得到的固体产物与第三部分钛化合物进行第五接触反应,得到所述固体催化剂组分;
44.所述助析出剂包括助析出剂a、助析出剂b和助析出剂c,所述助析出剂a为式(i)所示的二醇酯化合物;
[0045][0046]
式(i)中,r
1-r2相同或不同,各自独立地为取代或未取代的直链的c
1-c
20
烷基、取代或未取代的支链c
3-c
20
烷基、取代或未取代的c
3-c
20
环烷基、取代或未取代的c
6-c
20
芳基、取
代或未取代的c
7-c
20
烷芳基、取代或未取代的c
7-c
20
芳烷基、取代或未取代的c
2-c
10
烯烃基或取代或未取代的c
10-c
20
稠环芳基;r
3-r8相同或不同,各自分别为氢、卤素、取代或未取代的直链的c
1-c
20
烷基、取代或未取代的支链c
3-c
20
烷基、取代或未取代的c
3-c
20
环烷基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
7-c
20
烷芳基、取代或未取代的c
7-c
20
芳烷基、取代或未取代的c
2-c
10
烯烃基或者取代或未取代的c
10-c
20
稠环芳基,或者r
3-r6中的至少一个与r
7-r8中的至少一个成环;
[0047]
所述助析出剂b为脂肪族羧酸和/或芳香族羧酸的烷基酯中的至少一种;
[0048]
所述助析出剂c为通式ti(or9)nx
4-n
所示的钛酸酯类化合物中的至少一种,其中,r9为c
1-c
10
的烷基或c
3-c
10
的环烷基,x为卤素,1≤n≤4,n为整数;
[0049]
所述内给电子体化合物d为式(ii)所示的1,3-二醚类化合物中的至少一种,
[0050][0051]
式(ii)中r1’
和r2’
相同或不同,各自独立地为c
1-c
10
的直链或支链烷基;r4’
和r5’
相同或不同,各自独立地为c
1-c
20
的直链或支链烷基、c
3-c
20
的环烷基、c
6-c
20
的取代或未取代的芳基和c
7-c
20
的取代或未取代的烷芳基中的一种,且r4’
和r5’
可选择地相互键合生成环状结构;r3’
和r6’
相同或不同,各自独立地为氢和c
1-c
10
的直链或支链烷基中的一种。
[0052]
本发明的发明人发现,在z-n聚烯烃催化剂中,作为内给电子体的化合物和作为聚合活性中心的钛均吸附于活性氯化镁的晶格表面,一定程度上存在竞争吸附关系。换言之,内给电子体化合物在氯化镁表面的吸附量和状态会影响到作为活性中心的钛的数量和质量。另外,研究表明,作为活性中心的钛的总量并非越高越好,作为本发明所期望的用于纤维用聚烯烃制备的催化剂组分,希望钛含量控制在1.5-2.5重量%的理想范围。如果钛含量过高,催化剂更易在应用于连续聚合时出现温度波动等不稳定不可控现象;并且,钛含量过高,催化剂中的低定向能力的活性中心增多,会降低最终纤维用聚烯烃产品的立构规整性,从而降低产品性能。另外,基于1,3-二醚类化合物高昂的制备成本考虑,必须要提高其使用效率。因此,尤其是使用含有1,3-二醚类化合物和二醇酯化合物的催化剂组分,同时控制催化剂中钛原子含量对于制备具有窄分布、高等规指数的用作纤维料的烯烃聚合物有重要的影响。
[0053]
本发明中,通过使用1,3-二醚类化合物作内给电子体化合物d,并严格控制1,3-二醚类化合物与作为助析出剂a的二醇酯类化合物的用量比以及按特定比例分步加入内给电子体化合物d,从而制备了综合性能更加优良的、适于制备纤维用聚烯烃的催化剂固体组分。该催化剂固体组分的颗粒形态良好、聚合活性更高、定向能力更高、获得的聚烯烃分子量分布更窄。
[0054]
根据本发明,所述制备方法中,所述醇类化合物、钛化合物、内给电子体化合物d与所述卤化镁的用量,可以根据预期的纤维用聚烯烃的催化剂组分的组成进行适当地选择。优选情况下,所述钛化合物以钛元素计,所述卤化镁以镁元素计,所述醇类化合物、钛化合物的总量、内给电子体化合物d的总量与所述卤化镁的摩尔比可以为2-4:12-160:0.01-3:
1;优选为2.5-3.5:20-140:0.02-0.3:1。
[0055]
本发明提供的方法中,钛化合物可以分多次加入,如步骤(2)中加入第一部分钛化合物,步骤(4)中加入第二部分钛化合物,步骤(5)中加入第三部分钛化合物等。该方法还可以根据需要再增加加入钛化合物的步骤,直到满足制备方法需要提供的钛化合物的总量。另外第一部分钛化合物的加入量可以满足第一部分钛化合物:其与所述卤化镁(以镁元素计)的摩尔比为3-40:1,优选为5-35:1;加入的其它部分钛化合物的量可以只要使最终钛化合物的加入总量符合前述与所述卤化镁之间的摩尔比即可。分步加入钛化合物可以更好地控制钛活性组分在催化剂组分中的总量控制和良好分布,提高内给电子体化合物d的使用效率,有利于得到具有更窄的分子量分布和较高的链规整性的纤维用聚烯烃。
[0056]
根据本发明,所述制备方法的步骤(1)中,所述第一接触反应的目的是为了使所述卤化镁、醇类化合物在烃类溶剂存在下形成卤化镁与醇的加合物(又可称为卤化镁醇合物)的均匀溶液。本发明对进行第一接触反应以形成均匀溶液的条件没有具体的限定,可以根据使用的具体卤化镁化合物而确定。优选情况下,形成均匀溶液的第一接触反应可以在醇合条件下进行,所述第一接触反应的条件通常可以包括:反应温度为30-150℃,优选为60-140℃;反应时间为0.5-10小时,优选为0.5-6小时。
[0057]
根据本发明,所述卤化镁可以为二卤化镁、二卤化镁的水或醇的络合物、二卤化镁分子式中的一个或两个卤原子被烃基或卤烃氧基所置换的衍生物的一种或多种。本发明中,所述卤素可以为氟、氯、溴和碘中的至少一种,优选为氯和/或溴。所述卤化镁的具体实例可以为二氯化镁、二溴化镁、氯化苯氧基镁、氯化异丙氧基镁、氯化丁氧基中的一种或多种,优选为无水二氯化镁。
[0058]
根据本发明,所述醇类化合物可以为脂肪醇、脂环醇与芳香醇中的至少一种。其中脂肪醇优选为c
1-c
10
的直链或c
3-c
10
的支链脂肪醇;脂环醇优选为c
3-c
12
的脂环醇;芳香醇优选为c
6-c
20
的芳基醇或c
7-c
20
的烷基芳基醇。所述醇类化合物具体的实例可以为乙醇、丙醇、丁醇、2-乙基己醇、苯甲醇和苯乙醇中的至少一种,优选为2-乙基己醇。
[0059]
根据本发明,制备方法的步骤(1)中,使用的所述烃类溶剂可以为本领域常用的各种不与所述卤化镁醇合物发生化学相互作用的烃类溶剂,具体的实例可以为直链或支链烷烃、环烷烃、芳香烃的一种或多种,优选为癸烷、苯、甲苯、二甲苯中的一种或多种,更优选为甲苯。
[0060]
根据本发明,所述制备方法的步骤(2)的具体操作中没有特别限制,可参照现有技术进行,例如步骤(2)中的第一部分钛化合物可以单独与步骤(1)所得的均匀溶液在助析出剂的存在下进行反应,也可以将第一部分钛化合物与惰性稀释剂进行混合后与所述均匀溶液在助析出剂的存在下进行反应。优选情况下,将第一部分钛化合物与惰性稀释剂进行混合后与所述均匀溶液在助析出剂的存在下进行第二接触反应。一般地,根据本发明的所述制备方法,所述惰性稀释剂可以为己烷、庚烷、辛烷、癸烷、苯、甲苯和二甲苯中的至少一种。第一部分钛化合物(以钛元素计)与所述惰性稀释剂的摩尔比可以为1-20:1,优选为2-8:1。
[0061]
根据本发明,步骤(2)中的所述第二接触反应的条件可以包括:在助析出剂的存在下,将步骤(1)所得的均匀溶液与第一部分钛化合物在-40℃至0℃温度下接触3-5小时,然后升温至50-150℃;优选地,在助析出剂的存在下,将步骤(1)所得的均匀溶液与第一部分钛化合物在-30℃至-20℃温度下接触3.5-4.5小时,然后升温至90-130℃。步骤(2)中,可以
在-40℃至0℃温度下,先将所述钛化合物与所述惰性稀释剂的混合物和所述均匀溶液混合,然后加入助析出剂溶液接触3-5小时,然后升温至50-150℃,得到含固体沉淀的混合物;或者先将助析出剂溶液加入到所述均匀溶液中,然后在-40℃至0℃温度下,再与所述钛化合物与所述惰性稀释剂的混合物接触3-5小时后升温至50-150℃,得到含固体沉淀的混合物;优选先将助析出剂溶液加入到所述均匀溶液中,然后在-40℃至0℃温度下,与所述钛化合物与所述惰性稀释剂的混合物接触3-5小时后升温至50-150℃,得到含固体沉淀的混合物;更优选为先将助析出剂溶液加入到所述均匀溶液中,然后在-30℃至-20℃温度下,与所述钛化合物与所述惰性稀释剂的混合物接触3.5-4.5小时后升温至90-130℃,得到含固体沉淀的混合物。
[0062]
根据本发明,步骤(2)中,所述助析出剂的总用量可以是,所述卤化镁(以镁元素计)与所述助析出剂的总摩尔比为1:0.025-0.9,优选为1:0.03-0.3。
[0063]
根据本发明,优选情况下,所述卤化镁以镁元素计,步骤(2)中,所述卤化镁与所述助析出剂a的摩尔比为1:0.005-0.1,优选为1:0.01-0.05。优选地,所述卤化镁与所述助析出剂b的摩尔比为1:0.01-0.5,更优选为1:0.02-0.2。优选地,所述卤化镁与所述助析出剂c的摩尔比为1:0.01-0.3,更优选为1:0.02-0.08。
[0064]
根据本发明,在多个步骤中分步加入的钛化合物可以为本领域常规使用的各种钛化合物,优选情况下,所述钛化合物可以为三卤化钛和/或通式ti(or
10
)mx
4-m
所示的钛化合物,该通式中,r
10
可以为烷基,优选为c
1-c
10
的烷基,x可以为卤素,如cl、br或i,0≤m≤3,m为整数。优选地,所述钛化合物为四氯化钛、四溴化钛、四碘化钛、烷氧基三卤化钛、二烷氧基二卤化钛、三烷氧基卤化钛中的一种或多种,优选为四氯化钛。
[0065]
根据本发明,优选情况下,所述助析出剂a可以为2-乙基-1,3-丙二醇二苯甲酸酯、2-丙基-1,3-丙二醇二苯甲酸酯、2-异丙基-2-异戊基-1,3丙二醇二苯甲酸酯、1,3-丁二醇二甲基苯甲酸酯、2-甲基-1,3-丁二醇二间氯苯甲酸酯、2,3-二甲基-1,3-丁二醇二苯甲酸酯、1,3-戊二醇新戊酸酯、2,4-戊二醇二苯甲酸酯、2-甲基-1,3-戊二醇苯甲酸肉桂酸酯、2,2-二甲基-1,3-戊二醇二苯甲酸酯、2,4-庚二醇二苯甲酸酯、3,5-庚二醇二苯甲酸酯、4-乙基-3,5-庚二醇二苯甲酸酯和2-甲基-3,5-庚二醇二苯甲酸酯中的至少一种;优选为3,5-庚二醇二苯甲酸酯、4-乙基-3,5-庚二醇二苯甲酸酯和2,4-戊二醇二苯甲酸酯中的至少一种;更优选为3,5-庚二醇二苯甲酸酯。
[0066]
根据本发明,优选地,所述助析出剂b中的脂肪族羧酸的碳原子数优选为1-8,芳香族羧酸的碳原子数优选为7-10,烷基的碳原子数优选为1-10。优选地,所述助析出剂b可以为苯甲酸乙酯、邻苯二甲酸二乙酯、邻苯二甲酸二正丁酯、邻苯二甲酸二异丁酯、邻苯二甲酸二异辛酯、邻苯二甲酸二正辛酯、己二酸二乙酯和己二酸二丁酯中的至少一种;更优选为苯甲酸烷基酯、邻苯二甲酸二烷基酯和己二酸二烷基酯中的至少一种;进一步优选为邻苯二甲酸二正丁酯和/或邻苯二甲酸二异丁酯。
[0067]
根据本发明一种优选实施方式,所述助析出剂a与所述助析出剂b不同。
[0068]
根据本发明,优选地,所述助析出剂c中的r9的碳原子数优选为2-6;更优选,所述助析出剂c为钛酸四丁酯、钛酸四乙酯和钛酸四异丙酯中的至少一种;进一步优选为钛酸四丁酯。
[0069]
根据本发明,在步骤(3)中,为了制备分子量分布更窄且规整性更高的烯烃聚合
物,第一部分内给电子体化合物d优选为式(ii)所示的1,3-二醚类化合物。其它业内所熟知的内给电子体化合物如式(i)所示的二元醇酯类化合物、邻苯二甲酸酯类、丙二酸酯类、琥珀酸酯类、戊二酸酯类、新戊酸酯或碳酸酯类化合物因制备的聚合物分子量分布宽,均不适于用作本发明内给电子体化合物d。
[0070]
根据本发明,所述制备方法的步骤(3)中,所述第三接触反应的条件包括:反应温度为20-120℃,优选为70-110℃;反应时间为0.5-6小时,优选为1-4小时。
[0071]
优选地,第一部分内电子体化合物d可以为2-异丙基-2-异戊基-1,3-二甲氧基丙烷、9,9-二(甲氧基甲基)芴、2-异丁基-2-异丙基-1,3-二甲氧基丙烷、2,2-二环戊基二甲氧基丙烷、2,2-二苯基-1,3-二甲氧基丙烷、2-异丁基-2-异丙基-1,3-二甲氧基丙烷、2,2-二环戊基-1,3-二甲氧基丙烷和2,2-二异丁基-1,3-二甲氧基丙烷中的至少一种;优选为2-异丙基-2-异戊基-1,3-二甲氧基丙烷和/或9,9-二(甲氧基甲基)芴。
[0072]
本发明中,第一部分内给电子体化合物d只能在步骤(2)中固体沉淀析出后加入,优选在步骤(3)中加入。这主要是因为,在步骤(1)或步骤(2)固体沉淀析出之前加入,一方面会影响到步骤(2)中固体沉淀的析出效果,即影响到催化剂组分的颗粒形态,并最终影响到催化剂的综合性能;另一方面,研究表明在固体沉淀析出之前加入内给电子体化合物d还会影响到内给电子体化合物d的使用效率,具体说,即便加入大量的1,3-二醚类化合物也很难在最终催化剂固体组分中获得较多的1,3-二醚类化合物,从而影响到最终制备树脂的性能,使其不适于纺粘用聚丙烯的制备的使用。
[0073]
根据本发明,所述制备方法的步骤(4)中,再次加入第二部分内给电子体化合物d。第二部分内给电子体化合物d可以是上述第一部分内给电子体化合物d,具体选择的化合物可以相同或不同。将内给电子体化合物d分步以特定比例在步骤(3)和(4)加入,且用量可以按上述限定。
[0074]
相对于如果在步骤(3)中加入所有的内给电子体化合物,采用在步骤(3)和步骤(4)中分别加入内给电子体化合物d的制备方法,可以在以下几个方面改善催化剂的综合性能:1、可以使负载于所述卤化镁的所述内给电子体化合物d更加均一,从而使得制备得到的催化剂的活性衰减有所改善。具体地,制备得到的催化剂的2小时的聚合活性与1小时的聚合活性的比值会增加,表明催化剂的活性衰减变慢。这是高性能催化剂所追求的目标;2、提高内给电子体化合物d的使用效率,即使用总量更少的内给电子体化合物d来使得催化剂固体组份中的内给电子体化合物d含量相同;3、有效降低催化剂中的钛元素的含量,从而减少不稳定活性中心以及低定向能力活性中心,降低催化剂在聚合反应过程中反应器温度波动风险,并避免生成的聚合物等规指数偏低的问题。以上三点对于制备用作纤维用的烯烃聚合物十分有益。
[0075]
本发明中,步骤(4)和(5)中钛化合物的使用形式没有特别限制,例如步骤(4)或(5)中的钛化合物可以单独地直接使用,也可以将钛化合物先与惰性稀释剂混合,然后再与所述固体组分进行第四接触反应,或者与所述固体产物进行所述第五接触反应。所述钛化合物和惰性稀释剂如前文所述,在此不再赘述。
[0076]
本发明中,步骤(4)和(5)中的所述第四接触反应和第五接触反应条件可以相同,包括:反应温度为50-150℃,优选为80-120℃;反应时间为1-6小时,优选为2.5-4.5小时。
[0077]
本发明提供的催化剂组分的制备方法还可以包括,完成步骤(5)后,过滤掉所述第
五接触反应得到的产物中的液体,得到固体反应产物,再重复所述第五接触反应1-3次;经过洗涤、干燥,得到固体的含钛的催化剂组分,以提供固体的催化剂组分作为用于烯烃聚合的催化剂组分。
[0078]
本发明的步骤(4)和(5)中,加入的第二部分钛化合物、第三部分钛化合物的量,只要满足使钛化合物的总量符合前述与所述卤化镁(以镁元素计)的摩尔比即可,例如可以第二部分钛化合物或第三部分钛化合物与卤化镁的摩尔比可以为3-40:1,优选为5-35:1。
[0079]
根据本发明的一种优选实施方式,本发明提供的催化剂固体组分的方法可以按照如下步骤进行:
[0080]
(1)在烃类溶剂下,将无水氯化镁和醇类化合物在30-150℃(优选为60-140℃)进行第一接触反应0.5-10小时(优选为0.5-6小时),得到均匀的醇合物溶液,其中,无水氯化镁与醇类化合物的摩尔比为1:2-4(优选为1:2.5-3.5);
[0081]
(2)将二醇酯类化合物(助析出剂a)、脂肪族或者芳香族的羧酸的酯类化合物(助析出剂b)和钛酸酯类化合物(助析出剂c)加入到上述醇合物溶液中,无水氯化镁与二醇酯类化合物的摩尔比为1:0.005-0.1(优选为1:0.01-0.05),无水氯化镁与酯类化合物的摩尔比为1:0.01-0.5(优选为1:0.02-0.2),无水氯化镁与钛酸酯类化合物的摩尔比为1:0.01-0.3(优选为1:0.02-0.08);
[0082]
将上述加有助析出剂的醇合物溶液,加入到-40℃至0℃的第一部分钛化合物与惰性稀释剂的混合物中,通过搅拌在-40℃至0℃(优选为-30℃至-20℃)下反应,并在3-5小时(优选为3.5-4.5小时)内升温至50-150℃(优选为90-130℃),升温过程中析出固体沉淀,完成第二接触反应,得到含固体沉淀的混合物;其中,第一部分钛化合物与无水氯化镁的摩尔比为3-40:1,优选为5-35:1;
[0083]
(3)在上述(2)中得到的混合物中加入第一部分内给电子体化合物d,在温度为20-120℃(优选为70-110℃)下进行第三接触反应0.5-6小时(优选为1-4小时),得到悬浮物,其中,二醇酯类化合物与加入的内给电子体化合物d的总量的摩尔比为0.05:1至小于0.5:1(优选为0.1-0.3:1);加入的第一部分内给电子体化合物d与后续步骤(4)中加入的第二部分内给电子体化合物d之间的摩尔比为0.1-10:1(优选为0.2-5:1,更优选0.2-1:1);
[0084]
(4)将上述(3)中得到的悬浮物进行固液分离过滤掉液体,得到固体组分,并将向固体组分中加入第二部分钛化合物与惰性稀释剂的混合物、第二部分内给电子体化合物d,在50-150℃(优选为80-120℃)下进行第四接触反应1-6小时(优选为2.5-4.5小时),反应结束后,过滤出液体,得到固体产物;其中,第二部分钛化合物与无水氯化镁的摩尔比为3-40:1,优选为5-35:1;
[0085]
(5)将上述(4)中得到的固体产物,与第三部分钛化合物与惰性稀释剂的混合物在50-150℃下(优选为80-120℃)进行第五接触反应1-6小时(优选为2.5-4.5小时),得到产物,其中,第三部分钛化合物与无水氯化镁的摩尔比为3-40:1,优选为5-35:1;
[0086]
(6)将上述(5)得到的产物,过滤出液体,得到固体反应产物,将钛化合物和惰性稀释剂的混合物与该固体反应产物重复进行所述第五接触反应1-3次,经过洗涤,干燥,得到固体的含钛的催化剂组分;其中,所述钛化合物与无水氯化镁的摩尔比可以为3-40:1,优选为5-35:1。
[0087]
本发明的一种优选实施方式中,特别优选助析出剂a为3,5-庚二醇二苯甲酸酯,内
给电子体化合物d为2-异丙基-2-异戊基-1,3-二甲氧基丙烷和/或9,9-二(甲氧基甲基)芴;所述助析出剂a与所述内给电子体化合物d的总量的摩尔比为0.05-0.5:1,优选为0.1-0.3:1;步骤(3)中,所述第一部分内给电子体化合物d与第二部分内给电子体化合物d之间的摩尔比为0.1-10:1,优选为0.2-5:1,更优选0.2-1:1。
[0088]
本技术中,可以特别地使用特定的助析出剂a、内给电子体化合物d,且分步加入内给电子体化合物d和钛化合物,且限定特定的用量,从而更好地解决本技术的技术问题。
[0089]
本发明的固体催化剂组分中,优选地,以催化剂组分的总量为基准,含有1-2.5重量%的钛,1-3.5重量%的二醇酯化合物,6-15重量%的1,3-二醚类化合物,且二醇酯化合物与1,3-二醚类化合物的摩尔比为0.05-0.5:1;更优选地,以催化剂组分的总量为基准,含有1.4-2.2重量%的钛,1.5-3重量%的二醇酯化合物,7-11重量%的1,3-二醚类化合物,且二醇酯化合物与1,3-二醚类化合物的摩尔比为0.1-0.3:1。
[0090]
本发明中,所述固体催化剂、有机铝以及外给电子体的用量可根据需要确定,优选地,所述固体催化剂组分与有机铝化合物以钛/铝摩尔比计的用量比为1:25-100。
[0091]
本发明中,所述有机铝化合物作为助催化剂,优选为烷基铝化合物,包括但不限于:三乙基铝、三异丁基铝、三正丁基铝、三正己基铝、一氯二乙基铝、一氯二异丁基铝、一氯二正丁基铝、一氯二正己基铝、二氯一乙基铝、二氯一异丁基铝、二氯一正丁基铝和二氯一正己基铝中的一种或多种。所述烷基铝化合物更优选为三烷基铝,如:三乙基铝、三异丁基铝、三正丁基铝。
[0092]
根据本发明,所述外给电子体优选选自甲基-环戊基-二甲氧基硅烷、甲基-异丙基-二甲氧基硅烷、异丙基-环戊基-二甲氧基硅烷、二吡啶基二甲氧基硅烷、双全氢异喹啉二甲氧基硅烷、乙基-环戊基-二甲氧基硅烷、正丙基-环戊基-二甲氧基硅烷、异丙基-环戊基-二甲氧基硅烷、二(2-甲基丁基)-二甲氧基硅烷、二(3-甲基丁基)-二甲氧基硅烷、2-甲基丁基-3-甲基丁基-二甲氧基硅烷、二(2,2-二甲基-丙基)-二甲氧基硅烷、2-甲基丁基-2,2-二甲基-丙基-二甲氧基硅烷、3-甲基丁基-2,2-二甲基-丙基-二甲氧基硅烷、二甲基-二甲氧基硅烷、二甲基-二乙氧基硅烷、二异丙基-二甲氧基硅烷、二异丙基-二乙氧基硅烷、二异丁基-二甲氧基硅烷、二异丁基-二乙氧基硅烷、甲基-环已基-二甲氧硅烷、甲基-异丁基-二甲氧基硅烷、二环已基-二甲氧基硅烷、二环已基-二乙氧基硅烷、二环戊基-二甲氧基硅烷、二环戊基-二乙氧基硅烷中的至少两种。
[0093]
优选地,所述外给电子体为甲基-环已基-二甲氧硅烷和二环戊基-二甲氧基硅烷以任意比例的混合物。优选地,以其中一种硅烷类化合物的摩尔质量为1mol计,其他硅烷类化合物的摩尔质量为0.2-5mol,优选为0.5-2mol。
[0094]
优选地,催化剂中的有机铝化合物与所述外给电子体的摩尔比以铝/硅计为1-60:1,优选为5-25:1。
[0095]
本发明提供的制备方法,可以是丙烯的均聚合,也可以是丙烯和其他烯烃的共聚合,优选为丙烯均聚合。其他烯烃为除丙烯外的c
2-c
12
烯烃,可以为乙烯或c
4-c
12
的α-烯烃,所述c
4-c
12
的α-烯烃的具体实例包括:1-正丁烯、1-正戊烯、1-正己烯、1-正辛烯和4-甲基-1-戊烯中的至少一种。优选地,所述除丙烯外的c
2-c
12
烯烃为乙烯或1-正丁烯。
[0096]
共聚合时,除丙烯外的c
2-c
12
烯烃可以为本领域常规选择或根据需要确定,本发明对此没有特别限定。
[0097]
作为外给电子体的硅烷类化合物可以在两个以上串联操作反应器内一并加入也可以分别加入,可以直接加入到反应器内,也可加到反应器进料相关的设备或管线上。
[0098]
根据本发明,在用于烯烃聚合的催化剂的制备过程中,有机铝化合物和任选的外给电子体可以分别与用于烯烃聚合的催化剂组分混合后反应,或者也可以将有机铝化合物和任选的外给电子体事先混合后再与用于烯烃聚合的催化剂组分混合并反应。
[0099]
本发明所述的催化剂可以直接加入到反应器内,也可以经过业界共知的预络合和/或预聚合之后,再加入到反应器内。
[0100]
所述预络合过程可以在有或无聚合单体的环境,如预络合或聚合反应器内进行。当单独进行预络合反应时,反应器的形式可以是连续搅拌釜反应器,也可以是能获得充分混合效果的其它形式,如环管反应器、含静态混合器的一段管路,甚至也可以是一段物料处于湍流状态的管路。预络合的温度可控制在-10℃至60℃之间,优选的温度为0-30℃。预络合的时间控制在0.1-180min,优选的时间为5-30min。
[0101]
经过或不经过预络合的催化剂还可以进行任选地进行预聚合处理。预聚合可在液相本体条件下连续进行,也可以在惰性溶剂中间歇进行。预聚合反应器可以是连续搅拌釜、环管反应器等。预聚合的温度可控制在-10℃至60℃之间,优选的温度为0-40℃。预聚合的倍数控制在0.5-1000倍,优选的倍数为1.0-500倍。
[0102]
根据本发明,所述烯烃的聚合反应可以按照现有的方法进行,具体地,在惰性气体的保护下,在液相单体或含聚合单体的惰性溶剂中,或在气相中,或通过在气液相中的组合聚合工艺进行连续聚合反应。
[0103]
在烯烃的聚合反应过程中,所述惰性气体、溶剂的种类和用量为本领域技术人员公知,在此将不再赘述。
[0104]
根据本发明,所述聚合为液相聚合和/或气相聚合;
[0105]
液相聚合时,采用氢气作为分子量调节剂,聚合温度为0-150℃,优选为40-100℃;聚合压力高于丙烯在相应聚合温度下的饱和蒸汽压力。
[0106]
气相聚合时,聚合温度为0-150℃,优选为40-100℃;聚合压力大于等于常压,优选为0.5-2.5mpa。本发明的压力均指表压。
[0107]
聚合产物经湿氮气去除未反应的催化剂的活性,蒸汽出去聚合物中残存的烷烃和油脂,加热干燥,得到聚丙烯粉料。
[0108]
将聚丙烯粉料与复合助剂、过氧化物和气味吸附剂混合后,用双螺杆挤出机造粒。
[0109]
本发明还提供由上述制备方法制得的聚丙烯树脂。
[0110]
本发明的聚丙烯为纺粘纤维用聚丙烯,可以用于制备纤维或无纺布中,具体地,可用于家纺内衬、包装材料、汽车内饰、医疗防护和卫生用品,如口罩、防护服、纸尿裤、湿巾用非织造布。
[0111]
下面结合实施例对本发明作进一步说明,但本发明的范围并不局限于这些实施例。
[0112]
实施例中数据根据以下测试方法获得:
[0113]
催化剂组分中助析出剂a化合物含量(二醇酯)以及内给电子体化合物d(1,3-二醚)的含量:采用waters 600e液相色谱进行测定或agilent 7890气相色谱测定。
[0114]
熔融指数(mfr):根据astm d1238,在230℃、2.16kg载荷下测定。
[0115]
分子量及其分子量分布(mw/mn):采用英国polymer laboratories公司产pl-gpc220凝胶渗透色谱仪和西班牙polymer char公司产的ir4检测器联用测定样品的分子量及其分布,色谱柱为3根串联plgel 10μm mixed-b柱,溶剂及流动相为1,2,4-三氯苯,柱温150℃,流速1.0ml/min。
[0116]
聚丙烯等规度指数(ii):采用庚烷抽提法测定,即取2g干燥的聚合物样品,置于抽提器中用沸腾庚烷抽提6小时,之后将剩余物干燥至恒重,所得的聚合物重量(g)与2的比值即为等规度。
[0117]
聚合物的熔点和结晶温度采用示差扫描量热法(dsc)测试:将5mg样品置于坩埚中,在ta公司dsc25型差示扫描量热仪上测定。在氮气氛围下,以10℃/min的升温速度从25℃升温到200℃,保温5min以消除热历史,然后以10℃/min降至25℃,保温1min,最后以10℃/min升至200℃,记录第二次降温和第三次升温的扫描数据。
[0118]
气体挥发性有机物(voc)含量:按照德国汽车工业联合会的标准vda277测试。
[0119]
气味按照德国工业联合会的标准vda270测试。
[0120]
反应器内气体摩尔比例:用气相色谱法测定。
[0121]
二甲苯可溶物含量:按astm d5492-98测定。
[0122]
拉伸屈服应力:按照iso527-2(mpa)中规定的方法测定。
[0123]
弯曲模量:根据astm d790测量注塑样品。
[0124]
实施例1
[0125]
本实施例用于说明本发明的聚丙烯及其制备方法。所述聚丙烯以及聚丙烯树脂按以下方法获得:
[0126]
(1)在经过高纯氮重复置换的反应釜中,依次加入3.150mol(300.0g)的无水氯化镁、19.68mol(2.1l)的甲苯、8.4mol(1.1l)的2-乙基己醇,在搅拌转速450rpm、温度为110℃的条件下,反应3.0小时,得到稳定均匀的醇合物溶液;
[0127]
(2)向上述醇合物溶液中加入3,5-庚二醇二苯甲酸酯48mmol(24ml)、邻苯二甲酸二异丁酯336mmol(90ml)和钛酸四丁酯132mmol(45ml),搅拌60分钟,冷却至室温,得到均匀溶液;将上述均匀溶液加入到经氮气充分置换、并装有-20℃的60mol(6.6l)的四氯化钛及11.4mol(1.2l)的甲苯的反应器中,通过搅拌使它们在-20℃下充分接触,经5小时后,升温至100℃,升温过程中析出固体沉淀,得到含有固体沉淀的混合物;
[0128]
(3)将2-异丙基-2-异戊基-1,3-二甲氧基丙烷231mmol(50g)加入上述含有固体沉淀的混合物中反应1小时,反应结束后,过滤出液体;
[0129]
(4)然后将(3)中过滤得到的固体组分与40.8mol(4.32l)的甲苯、26.2mol(2.88l)的四氯化钛及323mmol(70g)的2-异丙基-2-异戊基-1,3-二甲氧基丙烷在100℃下接触1.5小时,反应结束后,过滤出液体;
[0130]
(5)然后将(4)中过滤得到的固体产物与40.8mol(4.32l)的甲苯及26.2mol(2.88l)的四氯化钛在110℃下接触反应0.5小时;
[0131]
(6)过滤(5)反应得到的产物,将过滤得到的固体反应产物用40.8mol(4.32l)甲苯及26.2mol(2.88l)四氯化钛在110℃下进行接触反应一次;将最终得到的固体物用55.14mol(7.2l)己烷洗涤5次,然后干燥,得到用于烯烃聚合的催化剂组分a1。
[0132]
聚合反应在一套聚丙烯中试装置上进行。
[0133]
聚合方法及步骤如下:
[0134]
预聚合:主催化剂cat-1、助催化剂(三乙基铝)以及外给电子体甲基-环已基-二甲氧硅烷(cmms)和二环戊基-二甲氧基硅烷(dcpms)的混合物(二者摩尔质量比为1:1),经10℃、20min预接触反应后,连续地加入预聚反应器进行预聚合反应器,三乙基铝(teal)流量为6g/hr,外给电子体流量为1.02g/hr,主催化剂流量为0.36g/hr。预聚合在丙烯液相本体环境下进行,温度为15℃,停留时间约为4min。
[0135]
预聚后催化剂连续地进入环管反应器中,在环管反应器内完成丙烯均聚反应,环管聚合反应温度70℃,反应压力为4.0mpa,环管反应器的进料中加入氢气,在线色谱检测的氢气浓度为0.04mol%。
[0136]
反应得到的聚合物经脱气、湿氮气去活处理后,得到聚合物产品。
[0137]
将聚合得到的粉料中加入抗氧剂(双(3,5-二叔丁基-4-羟基苄基膦酸单乙酯)钙和(2,4-二叔丁基苯基)亚磷酸三酯,质量比为1:1)1600ppm、硬脂酸钙400ppm、滑石粉(目数为3000)300ppm和过氧化物(过氧化二叔丁基和双(叔丁基过氧化异丙基)苯的混合物,二者重量比为1:1)600ppm,以及气味吸附剂(jh100a,宁波嘉和公司)3000ppm,用双螺杆挤出机造粒。注塑机制备符合gb标准的注塑样品,并测定其物理性质。测定结果如表2所示。
[0138]
实施例2
[0139]
实施例2所使用的主催化剂、助催化剂、外给电子体和聚合工艺条件与实施例1相同。与实施例1不同之处在于:环管反应器内氢气浓度为0.06mol%。具体工艺条件如表1所示,性能测定结果如表2所示。
[0140]
实施例3
[0141]
实施例3所使用的主催化剂、助催化剂、外给电子体和聚合工艺条件与实施例1相同。与实施例1不同之处在于:造粒过程中添加800ppm过氧化物。具体工艺条件如表1所示,性能测定结果如表2所示。
[0142]
实施例4
[0143]
实施例4所使用的主催化剂、助催化剂、外给电子体和聚合工艺条件与实施例1相同。与实施例1不同之处在于:外给电子体甲基-环已基-二甲氧硅烷(cmms)和二环戊基-二甲氧基硅烷(dcpms)的混合比例为2:1,外给电子体总量与实施例1相同。具体工艺条件如表1所示,性能测定结果如表2所示。
[0144]
实施例5
[0145]
实施例5所使用的主催化剂、助催化剂、外给电子体和聚合工艺条件与实施例1相同。与实施例1不同之处在于:外给电子体甲基-环已基-二甲氧硅烷(cmms)和二环戊基-二甲氧基硅烷(dcpms)的混合比例为1:2,外给电子体总量与实施例1相同。具体工艺条件如表1所示,性能测定结果如表2所示。
[0146]
实施例6
[0147]
实施例6所使用的主催化剂、助催化剂、外给电子体和预聚合步骤与实施例1相同。
[0148]
将预聚合后的催化剂连续地通入环管反应器中,在环管反应器内完成丙烯与乙烯的无规共聚反应,环管聚合反应温度为70℃,反应压力4.0mpa,环管反应器的进料中加入氢气和乙烯,在线色谱检测的氢气浓度为0.05mol%,乙烯浓度为1.1mol%。
[0149]
反应得到的聚合物经脱气、湿氮气去活处理后,得到聚合物产品。将聚合得到的粉
料中加入抗氧剂(双(3,5-二叔丁基-4-羟基苄基膦酸单乙酯)钙和(2,4-二叔丁基苯基)亚磷酸三酯,质量比为1:1)1600ppm、硬脂酸钙400ppm、滑石粉(目数为3000)300ppm和过氧化物(过氧化二叔丁基和双(叔丁基过氧化异丙基)苯的混合物,二者重量比为1:1)600ppm,气味吸附剂(jh100a,宁波嘉和公司)3000ppm以及成核剂(hyperform hpn-715)1000ppm,用双螺杆挤出机造粒。注塑机制备符合gb标准的注塑样品,并测定其物理性质。测定结果如表2所示。
[0150]
实施例7
[0151]
实施例7所使用的主催化剂、助催化剂、外给电子体和预聚合步骤与实施例1相同。
[0152]
将预聚合后的催化剂连续地通入两个串联的环管反应器中,在环管反应器内完成聚合反应。两环管的聚合反应温度为70℃,反应压力4.0mpa。控制环管反应器的工艺条件,使第一、第二环管的产率比为约55:45。在第一环管反应器进料中不加入氢气,在线色谱检测的氢气浓度《10ppmv,加入1-丁烯,浓度为6mol%。在第二环管反应器中加入一定量的氢气和1-丁烯,在线色谱检测的氢气浓度为0.04mol%,1-丁烯浓度为5mol%。
[0153]
反应得到的聚合物经脱气、湿氮气去活处理后,得到聚合物产品。
[0154]
将聚合得到的粉料中加入抗氧剂(双(3,5-二叔丁基-4-羟基苄基膦酸单乙酯)钙和(2,4-二叔丁基苯基)亚磷酸三酯,质量比为1:1)1600ppm、硬脂酸钙400ppm、滑石粉(目数为3000)300ppm和过氧化物(过氧化二叔丁基和双(叔丁基过氧化异丙基)苯的混合物,二者重量比为1:1)600ppm,气味吸附剂(jh100a,宁波嘉和公司)3000ppm以及成核剂(hyperform hpn-715)1000ppm,用双螺杆挤出机造粒。注塑机制备符合gb标准的注塑样品,并测定其物理性质。测定结果如表2所示。
[0155]
对比例1
[0156]
对比例1所使用的助催化剂、外给电子体和聚合工艺条件与实施例1相同。与实施例1不同之处在于:主催化剂(含钛的活性固体催化剂组分)采用中国专利cn93102795中实施例1描述的方法得到,其ti含量:2.2重量%,mg含量18.0重量%,邻苯二甲酸二异丁酯含量:11.2重量%;环管反应器内氢气浓度为0.13mol%。具体工艺条件如表1所示,性能测定结果如表2所示。
[0157]
对比例2
[0158]
对比例2所使用的主催化剂、助催化剂和聚合工艺条件与实施例1相同。与实施例1不同之处在于:所用外给电子体为二异丙基-二甲氧基硅烷(dipms),外给电子体总量与实施例1相同。具体工艺条件如表1所示,性能测定结果如表2所示。
[0159]
对比例3
[0160]
对比例3所使用的主催化剂、助催化剂、外给电子体和聚合工艺条件与实施例1相同。与实施例1不同之处在于:不使用气味吸附剂。具体工艺条件如表1所示,性能测定结果如表2所示。
[0161]
[0162][0163]
由表2数据可以看出,本发明得到的聚丙烯树脂具有更低的voc,更低的气味,同时具有更高的流动性。
[0164]
以上已经描述了本发明的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。
[0165]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1