一种同质非均相聚酰亚胺薄膜及其制备方法和应用与流程
1.本发明涉及聚酰亚胺领域,具体地说,是涉及适用于高频通讯技术的一种同质非均相聚酰亚胺薄膜及其制备方法和应用。
背景技术:
2.随着5g时代的到来,超高速、超高数据密度的高频率通信技术将全面铺开,5g技术标准中的fr2频段更是达到了毫米波的范畴。确保如此高频率电磁波的高保真传输对于制造电子器件,尤其是天线等信号接收和传输组件的介电材料是极大的挑战。根据电磁学原理,电磁波的频率越大,其能量越高,穿透力越强的同时,穿透损失也更高,同时衍射能力也越弱。因此,为了保证毫米波信号的高保真传输,电子器件中的介电材料必须具有很低的介电常数和介电损耗,从而尽可能的减少信号在器件传输过程中的延迟和消耗。
3.聚酰亚胺(pi)是一类在分子主链上含有酰亚胺环结构的高分子,是目前综合性能最好的功能性高分子材料之一,也是少数几种能够满足微电子行业的高分子材料之一。一直以来,pi材料都是微电子行业最重要的基础材料之一,pi薄膜,黏胶等被广泛应用于微电子器件的制造与封装。然而随着5g技术的逐步应用与推广,传统pi基材存在介电常数、介电损耗以及吸湿性偏高的问题,已无法满足5g时代高频高速通信的应用要求,因此,急需开发新型低介电常数、低介电损耗、低吸湿率的新型pi材料以满足高频通讯的技术需求。
技术实现要素:
4.本发提供了一种同质非均相聚酰亚胺薄膜材料,具有低介电常数、低介电损耗、低吸湿率的特点,解决了目前传统聚酰亚胺材料由于介电性能不达标无法应用于5g高频通讯技术的问题。
5.本发明目的之一为提供一种同质非均相聚酰亚胺薄膜,所述聚酰亚胺薄膜内结晶主要沿着平行薄膜表面的方向取向,取向因子》0.4。
6.本发明所述薄膜由部分结晶的聚酰亚胺材料得到,晶粒沿着平行薄膜表面的方向高度取向。
7.本发明所述聚酰亚胺薄膜的吸湿率《0.5%。
8.同时,在10ghz频率条件下测试,所述聚酰亚胺薄膜的介电常数《3.0,介电损耗《0.005。
9.本发明所述同质非均相聚酰亚胺薄膜,由于结晶取向效应,聚酰亚胺薄膜材料内分子链段堆砌紧密,分子链间距下降,有效限制了水分子的渗透与吸附,材料吸湿率下降,同时由于晶粒取向增大了分子间相互作用,使得在外电场下分子片段的运动能力减弱,因而介电常数和介电损耗下降。
10.本发明所述聚酰亚胺薄膜,其聚酰亚胺具有式(i)所示结构:
[0011][0012]
其中,ar1选自以下结构中的至少一种,
[0013][0014][0015]r’1、r
’2、r
’4、r
’5独立为氢、甲基、三氟甲基、苯基,r
’3为氧原子、羰基、亚甲基、六氟异丙基、砜基;
[0016]
ar2选自以下结构中的至少一种,
[0017][0018]
[0019]
r1、r2、r3、r4、r5、r6、r7、r8独立为氢原子、氟原子、甲基、三氟甲基、三氟甲氧基、1,1,2-三氟-2-(全氟丙氧基)乙氧基;r9、r
10
同时为甲基或三氟甲基。
[0020]
本发明目的之二为提供所述聚酰亚胺薄膜的制备方法,包括将二胺单体和二酐单体聚合得到聚酰胺酸溶液,接着亚胺化形成聚酰亚胺薄膜,然后进行同步双向拉伸步骤和/或热处理退火步骤。
[0021]
以上制备方法中,所述二胺单体为芳香族二胺,优选自2,2'-双(三氟甲基)-4,4'-二氨基联苯、4,4'-二氨基-2,2'-二甲基联苯、4,4'-二氨基联苯、3,3'-双(三氟甲基)-4,4'-二氨基联苯、3,3'-双(甲基)-4,4'-二氨基联苯、对苯二胺、间苯二胺、2,3,5,6-四氟-1,4-苯二胺、4,5-二氟苯-1,2-二胺、2-氟-1,4-苯二胺、2,2',3,3',5,5',6,6'-八氟-[1,1'-双苯基]-4,4'-二胺、4,4'-二氨基-2,2'-双(三氟甲氧基)对二氨基联苯、2,2'-双(1,1,2-三氟-2-(全氟丙氧基)乙氧基)-[1,1'-双苯基]-4,4'-二胺、3,4'-二氨基二苯醚、4,4'-二氨基二苯醚、1,3-双(2-三氟甲基-4-氨基苯氧基)苯、1,3-双(3-三氟甲基-4-氨基苯氧基)苯、2,2'-双[4-(4-氨基苯氧基苯基)]丙烷、2,2-双[4-(4-氨基苯氧基)苯基]六氟丙烷、1,3-双(3'-氨基苯氧基)苯、1,3-双(4'-氨基苯氧基)苯、1,4-双(3'-氨基苯氧基)苯中的至少一种。
[0022]
所述二酐单体为芳香族四酸二酐,优选自均苯四甲酸二酐、3,3',4,4'-联苯四甲酸二酐、2,2'-二甲基-3,3',4,4'-联苯四甲酸二酐、2,2'-二(三氟甲基)-3,3',4,4'-联苯四甲酸二酐、2,2'-二苯基-3,3',4,4'-联苯四甲酸二酐、3,3',4,4'-二苯醚四甲酸二酐、3,3',4,4'-二苯酮四酸二酐、5,5'-亚甲基双(异苯并呋喃-1,3-二酮)、1,3-二氧-1,3-双异苯并呋喃-5-基1,3-二氧-1,3-双异苯并呋喃-5-羧酸酯、4,4'-(4,4'-异丙基二苯氧基)二酞酸酐、、4,4'-(4,4'-异丙基二苯氧基)双(邻苯二甲酸酐)中的至少一种。
[0023]
本发明可采用的一些二胺单体和二酐单体的具体结构如下:
[0024]
tfdb:2,2'-双(三氟甲基)-4,4'-二氨基联苯/2,2'-bis(trifluoromethyl)benzidine),cas:341-58-2。
[0025][0026]
dmdb:4,4'-二氨基-2,2'-二甲基联苯/2,2'-dimethylbenzidine 4,4'-diamino-2,2'-dimethylbiphenyl,cas:84-67-3。
[0027][0028]
bpa:4,4'-二氨基联苯/4,4'-diaminobiphenyl,cas:92-87-5。
[0029][0030]
pda:对苯二胺/p-phenylenediamine,cas:106-50-3。
[0031]
[0032]
mpda:间苯二胺/m-phenylenediamine,cas:108-45-2。
[0033][0034]
6fpda:2,3,5,6-四氟-1,4-苯二胺/2,3,5,6-tetrafluoro-1,4-phenylenediamine,cas:1198-64-7。
[0035][0036]
2fpda:4,5-二氟苯-1,2-二胺/2,5-difluorobenzene-1,4-diamine,cas:698-52-2。
[0037][0038]
fpda:2-氟-1,4-苯二胺/2-fluorobenzene-1,4-diamine,cas:14791-78-7。
[0039][0040]
3tfdb:3,3'-双(三氟甲基)-4,4'-二氨基联苯/3,3'-bis(trifluoromethyl)benzidine)。
[0041][0042]
3dmdb:3,3'-双(甲基)-4,4'-二氨基联苯/3,3'-bis(methyl)benzidine)。
[0043][0044]
8fbpa:2,2',3,3',5,5',6,6'-八氟-[1,1'-双苯基]-4,4'-二胺/2,2',3,3',5,5',6,6'-octafluoro-[1,1'-biphenyl]-4,4'-diamine。
[0045][0046]
tfmob:4,4'-二氨基-2,2'-双(三氟甲氧基)对二氨基联苯/2,2'-bis-trifluoromethoxy-biphenyl-4,4'-diamine,cas:147835-68-5。
[0047][0048]
tfpob:2,2'-双(1,1,2-三氟-2-(全氟丙氧基)乙氧基)-[1,1'-双苯基]-4,4'-二胺/2,2'-bis(1,1,2-trifluoro-2-(perfluoropropoxy)ethoxy)-[1,1'-biphenyl]-4,4'-diamine。
[0049][0050]
pmda:均苯四甲酸二酐/pyromellitic dianhydride,cas:89-32-7。
[0051][0052]
bpda:3,3’,4,4
’-
联苯四甲酸二酐/3,3’,4,4
’-
biphenyl tetracarboxylic diandhydride,cas:2420-87-3。
[0053][0054]
dmbpda:2,2
’-
二甲基-3,3’,4,4
’-
联苯四甲酸二酐/6,6'-dimethyl-[5,5'-biisobenzofuran]-1,1',3,3'-tetraone。
[0055][0056]
dfbpda:2,2
’-
二(三氟甲基)-3,3’,4,4
’-
联苯四甲酸二酐/6,6'-bis(trifluoromethyl)-[5,5'-biisobenzofuran]-1,1',3,3'-tetraone。
[0057][0058]
odpa:3,3',4,4'-二苯醚四甲酸二酐/4,4'-oxydiphthalic anhydride,cas:1823-59-2。
[0059][0060]
btda:3,3',4,4'-二苯酮四酸二酐/3,3',4,4'-benzophenonetetracarboxylic dianhydride,cas:2421-28-5。
[0061][0062]
bdda:5,5'-亚甲基双(异苯并呋喃-1,3-二酮)/5,5'-methylenebis(isobenzofuran-1,3-dione)。
[0063][0064]
beda:1,3-二氧-1,3-双异苯并呋喃-5-基1,3-二氧-1,3-双异苯并呋喃-5-羧酸酯/1,3-dioxo-1,3-dihydroisobenzofuran-5-yl 1,3-dioxo-1,3-dihydroisobenzofuran-5-carboxylate。
[0065][0066]
以上制备方法中,将所述二胺单体和二酐单体在极性非质子溶剂中进行聚合,二胺单体与二酐单体的总量占反应体系的5~20wt%,优选为7~17wt%。
[0067]
以上制备方法中,所述二酐单体与二胺单体的摩尔比为(0.970~0.997):1,优选为(0.985~0.995):1。
[0068]
以上制备方法中,所述极性非质子溶剂优选自n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、乙腈、六甲基磷酰胺、γ-丁内酯、四甲基砜、n,n-二甲基丙烯基脲丙
酮、1,3-二甲基-2-咪唑啉酮、吡啶、四氢呋喃、环丁砜、间甲酚、二甘醇二甲醚中的至少一种。
[0069]
以上制备方法中,聚合反应温度为0~70℃,优选为20~60℃。
[0070]
以上制备方法中,聚酰胺酸的亚胺化可以通过高温热亚胺化或者化学亚胺化的方法进行。
[0071]
在高温热亚胺化中,亚胺化温度优选为300~450℃,更优选为320~430℃。
[0072]
以上制备方法中,所述同步双向拉伸步骤的md和td方向的拉伸比均为1.05~1.35,优选为1.10~1.30;
[0073]
所述拉伸温度为所制备聚酰亚胺薄膜材料的玻璃化转变温度以下15℃至以上50℃,优选为所制备聚酰亚胺薄膜材料的玻璃化转变温度以下10℃至以上35℃。
[0074]
以上制备方法中,所述热处理退火步骤的温度为所制备聚酰亚胺薄膜材料的玻璃化转变温度至以上75℃,优选为玻璃化转变温度以上10℃至50℃。
[0075]
所述热处理退火处理时间为50~350s,优选为100s~300s。
[0076]
所述热处理退火时,所述聚酰亚胺薄膜可在双向恒定应力固定下进行,双向恒定应力主要起到固定作用,无特殊限定。
[0077]
本发明目的之三为提供所述聚酰亚胺薄膜或者所述制备方法得到的聚酰亚胺薄膜材料在高频通讯领域中的应用。
[0078]
应用时,通讯所用电磁波频率》0.45ghz。
[0079]
本技术所得同质非均相聚酰亚胺薄膜内结晶主要沿着平行薄膜表面的方向取向,取向因子》0.4,在10ghz频率条件下测试,介电常数《3.0,介电损耗《0.005,且吸湿率《0.5%,可用于适用于高频通讯技术的同质非均相聚酰亚胺薄膜材料的工业生产以及下游产品应用中。
附图说明
[0080]
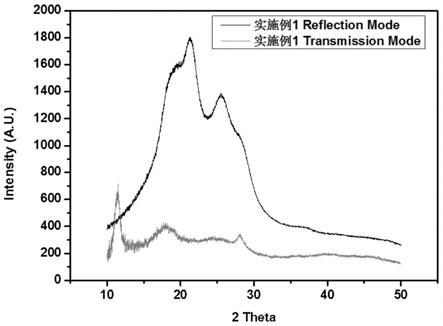
图1为实施例1聚酰亚胺薄膜材料xrd测试结果。
[0081]
图2为比较例1聚酰亚胺薄膜材料xrd测试结果。
[0082]
图3为实施例1聚酰亚胺薄膜材料xrd沿取向角积分结果。
[0083]
图4为比较例1聚酰亚胺薄膜材料xrd沿取向角积分结果。
具体实施方式
[0084]
下面结合具体实施例对本发明进行具体的描述,有必要在此指出的是以下实施例只用于对本发明的进一步说明,不能理解为对本发明保护范围的限制,本领域技术人员根据本发明内容对本发明做出的一些非本质的改进和调整仍属本发明的保护范围。
[0085]
本发明具体实施方式中所用原料为市售所得。
[0086]
聚酰亚胺薄膜的制备:
[0087]
首先以n,n-二甲基乙酰胺、n,n-二乙基乙酰胺、n,n-二甲基甲酰胺及n-甲基-2-吡咯烷酮等非质子溶剂作为反应介质,以二胺单体和二酐单体为反应物,形成聚酰胺酸溶液,反应温度为0~70℃,其中二胺和二酐重量占包括反应介质与反应物在内的总重量的5%~20%。聚酰胺酸的亚胺化可以通过高温热亚胺化或者化学亚胺化的方法实施。
[0088]
【实施例1】
[0089]
在三口烧瓶中加入26.66g(0.083mol)2,2'-双(三氟甲基)-4,4'-二氨基联苯(tfdb)和284g n-甲基-2-吡咯烷酮(nmp),搅拌溶解后加入21.82g(0.074mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)以及1.79g(0.0082mol)均苯四甲酸二酐(pmda),在60℃,氮气保护下搅拌反应48h,得到聚酰胺酸溶液。
[0090]
将上述聚酰胺酸溶液经过涂布以后首先在100℃、氮气保护的条件下脱除绝大部分溶剂,而后在氮气保护的条件逐步升温至450℃完成亚胺化,所得聚酰亚胺薄膜的玻璃化转变温度为363℃。所得到薄膜经过350℃、氮气保护的条件下同步双向拉伸(md拉伸倍率1.30,td拉伸倍率1.30)以后得到【实施例1】聚酰亚胺薄膜。
[0091]
【比较例1】
[0092]
聚酰胺酸溶液制备过程以及亚胺化过程与【实施例1】完全相同,但是没有同步双向拉伸步骤,得到【比较例1】聚酰亚胺薄膜。
[0093]
【实施例2】
[0094]
在三口烧瓶中加入12.74g(0.060mol)4,4'-二氨基-2,2'-二甲基联苯(dmdb)、5.06g(0.028mol)2,3,5,6-四氟-1,4-苯二胺(6fpda)以及340g n,n-二甲基乙酰胺(dmac),搅拌溶解后加入28.21g(0.08756mol)3,3',4,4'-二苯酮四酸二酐(btda),在45℃,氮气保护下搅拌反应24h,得到聚酰胺酸溶液。
[0095]
将上述聚酰胺酸溶液经过涂布以后首先在100℃、氮气保护的条件下脱除绝大部分溶剂,而后在氮气保护的条件逐步升温至320℃完成亚胺化,所得聚酰亚胺薄膜的玻璃化转变温度为351℃。所得到薄膜经过360℃、氮气保护的条件下退火300s(md恒定应力1.0mpa,td恒定应力1.0mpa)以后得到【实施例2】聚酰亚胺薄膜。
[0096]
【比较例2-1】
[0097]
聚酰胺酸溶液制备过程与【实施例2】完全相同,但是未经过退火步骤,经过涂布以及亚胺化以后得到【比较例2-1】聚酰亚胺薄膜。
[0098]
【比较例2-2】
[0099]
聚酰胺酸溶液制备过程以及亚胺化条件与【实施例2】完全相同,所得到薄膜经过340℃、氮气保护的条件下退火300s(md恒定应力1.0mpa,td恒定应力1.0mpa)以后得到【比较例2-2】聚酰亚胺薄膜。
[0100]
【比较例2-3】
[0101]
聚酰胺酸溶液制备过程以及亚胺化条件与【实施例2】完全相同,所得到薄膜经过430℃、氮气保护的条件下退火300s(md恒定应力1.0mpa,td恒定应力1.0mpa)以后得到【比较例2-3】聚酰亚胺薄膜。
[0102]
【实施例3】
[0103]
在三口烧瓶中加入16.01g(0.050mol)2,2'-双(三氟甲基)-4,4'-二氨基联苯(tfdb)和286g n,n-二甲基乙酰胺(dmac),搅拌溶解后加入9.60g(0.044mol)均苯四甲酸二酐(pmda)以及2.71g(0.0052mol)4,4'-(4,4'-异丙基二苯氧基)双(邻苯二甲酸酐)(bpada),在20℃,氮气保护下搅拌反应48h,得到聚酰胺酸溶液。
[0104]
将上述聚酰胺酸溶液经过涂布以后首先在100℃、氮气保护的条件下脱除绝大部分溶剂,而后在氮气保护的条件逐步升温至400℃完成亚胺化,所得聚酰亚胺薄膜的玻璃化
转变温度为360℃。所得到薄膜经过390℃、氮气保护的条件下同步双向拉伸(md拉伸倍率1.10,td拉伸倍率1.10),以后得到【实施例3】聚酰亚胺薄膜。
[0105]
【比较例3-1】
[0106]
聚酰胺酸溶液制备过程与【实施例3】完全相同,但是未经过同步双向拉伸步骤,经过涂布以及亚胺化以后得到【比较例3-1】聚酰亚胺薄膜。
[0107]
【比较例3-2】
[0108]
聚酰胺酸溶液制备过程与【实施例3】完全相同,经过涂布以及亚胺化以后所得到薄膜经过335℃、氮气保护的条件下同步双向拉伸(md拉伸倍率1.10,td拉伸倍率1.10),得到【比较例3-2】聚酰亚胺薄膜。
[0109]
【比较例3-3】
[0110]
聚酰胺酸溶液制备过程与【实施例3】完全相同,经过涂布以及亚胺化以后所得到薄膜经过420℃、氮气保护的条件下同步双向拉伸(md拉伸倍率1.10,td拉伸倍率1.10),得到【比较例3-3】聚酰亚胺薄膜。
[0111]
【实施例4】
[0112]
在三口烧瓶中加入8.01g(0.025mol)2,2'-双(三氟甲基)-4,4'-二氨基联苯(tfdb)、7.43g(0.035mol)4,4'-二氨基-2,2'-二甲基联苯(dmdb)和241g n,n-二甲基乙酰胺(dmac),搅拌溶解后加入搅拌溶解后加入17.48g(0.0594mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),在35℃,氮气保护下搅拌反应48h,得到聚酰胺酸溶液。
[0113]
将上述聚酰胺酸溶液经过涂布以后首先在100℃、氮气保护的条件下脱除绝大部分溶剂,而后在氮气保护的条件逐步升温至350℃完成亚胺化,所得聚酰亚胺薄膜的玻璃化转变温度为375℃。所得到薄膜首先经过370℃、氮气保护的条件下同步双向拉伸(md拉伸倍率1.30,td拉伸倍率1.30),而后经过390℃、氮气保护的条件下退火100s(md恒定应力1.0mpa,td恒定应力1.0mpa)以后得到【实施例4】聚酰亚胺薄膜。
[0114]
【实施例5】
[0115]
聚酰胺酸溶液制备以及亚胺化过程与【实施例4】完全相同,经过涂布以及亚胺化以后所得到薄膜首先经过390℃、氮气保护的条件下退火100s(md恒定应力1.0mpa,td恒定应力1.0mpa),而后经过370℃、氮气保护的条件下同步双向拉伸(md拉伸倍率1.30,td拉伸倍率1.30)以后得到【实施例5】聚酰亚胺薄膜。
[0116]
所得到上述聚酰亚胺薄膜的热膨胀系数(cte)、玻璃化转变温度、结晶度、晶粒取向、介电常数、介电损耗以及吸湿率通过如下方法测定:
[0117]
cte:采用热机械分析仪,在负载0.05n,升温速率10℃/分钟条件下对样品进行测试,通过对样品在50℃到200℃范围内尺寸变化平均值的计算得到cte。
[0118]
x射线衍射测试:分别通过掠入射x射线衍射(反射模式)和透射x射线衍射(投射模式)对所制备的聚酰亚胺薄膜进行测试。
[0119]
结晶度:通过如下方程计算,
[0120]
cr(%)=sc/(sc+sa)
[0121]
其中cr为百分率结晶度;sc为结晶峰积分面积;sa为无定形峰积分面积。sc与sa)通过origin软件对x射线衍射数据拟合分蜂得到。
[0122]
晶粒取向:通过分析反射模式与透射模式所获得的x射线衍射数据,评判不同晶面
在膜平面内以及垂直膜平面方向的分布情况,从而获得晶粒取向情况。
[0123]
取向因子:为了进一步定量表征晶粒取向程度,通过以下公式计算样品取向因子,
[0124][0125]
其中f为取向因子,为取向参数,
[0126][0127]
其中为方位角,为特定方位角xrd衍射强度。
[0128]
介电常数和介电损耗:根据ipc-tm-650-2.5.5.9标准,采用谐振腔法,在10ghz的条件下对所制备的聚酰亚胺薄膜材料进行测试。
[0129]
吸湿率:根据ipc-tm-650-2.6.2标准对所制备的聚酰亚胺薄膜进行吸湿率测试。
[0130]
聚酰亚胺薄膜的玻璃化转变温度的测定:采用热机械分析仪,在负载0.05n,升温速率10℃/分钟条件下对样品进行测试。
[0131]
通过上述方法对各实施例和比较例进行测试之后的结果总结于表1。
[0132]
表1各实施例和比较例测试结果
[0133][0134]
通过表1数据可以看出,晶粒取向对于降低材料介电常数、介电损耗以及吸湿率具有巨大作用。与晶粒未取向的比较例相比,具备晶粒取向特征的实施例均具有《3.0的介电常数、《0.005的介电损耗以及《0.5%的吸湿率。这主要是由于晶粒取向之后分子链堆砌变得紧密,分子链间距下降,因而不易被水分子所渗透。同时由于晶粒取向增大了分子间相互
作用,使得在外电场下分子片段的运动能力减弱,因而介电常数和介电损耗下降。
[0135]
同时,通过比较表1中实施例1、2、3与比较例1、2-1、3-1的测试结果可知,通过同步双向拉伸,高温退火等后处理方式可以提高聚酰亚胺薄膜的结晶度和晶粒取向,从而大幅改善材料的介电性能和吸湿率。这可能是由于在高温条件下,分子链段活动能力增强,结晶趋势变大,同时在外力的作用下,晶粒选择性的沿着外力作用方向发生取向。通过比较表1中实施例2与比较例2-2、2-3以及实施例3与比较例3-2、3-3的测试结果可知,后处理过程温度的选择对于最终材料性能的提升具有关键作用。无论退火或拉伸温度过高或是过低,均无法达到有效提升材料性能的目的。
[0136]
另外,由图1和图2可知,在没有经过同步双向拉伸或高温退火等后处理的条件下,比较例1聚酰亚胺薄膜结晶很弱,没有任何可观测到的晶粒取向。经过了双向拉伸之后,实施例1的结晶度大幅度提高,同时可以观测到xrd测试中反射模式与透射模式出现了不同位置和强度的衍射图谱,这是由于xrd反射模式主要观测到的是垂直薄膜表面方向的结晶与分子排列,而透射模式主要观测到的是平行薄膜表面方向的结晶与排列。经过分析得知,实施例聚酰亚胺薄膜材料在反射模式下探测到的衍射峰主要由(hk0)晶面贡献,透射模式下探测到的衍射峰主要由(00l)晶面贡献,因此证明了实施例聚酰亚胺薄膜材料结晶主要沿着平行于薄膜表面方向内取向。
[0137]
同时,分别对实施例与比较例反射模式下的xrd数据沿方位角进行积分,结果如图3和图4所示。相较于比较例,实施例1在0度左右方位角位置具有明显高于其他方位角位置的衍射强度,证明了实施例1聚酰亚胺薄膜材料中大部分结晶与膜平面平行,具有明显的结晶取向。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1