一种制备N-甲基-2-吡咯烷乙胺或其盐的方法与流程
一种制备n-甲基-2-吡咯烷乙胺或其盐的方法
技术领域
1.本发明涉及药物化学合成领域,具体涉及n-甲基-2-吡咯烷乙胺及其盐酸盐的制备方法,以及包括该方法的制备tpn729的方法。
背景技术:
2.tpn729,化学名为1-甲基-5-{2-丙氧基-5-[[1-甲基-1-(2-吡咯烷-1-基) 乙基]氨基磺酸基]苯基}-3-丙基-1,6-二氢-7h-吡唑并[4,3-d]嘧啶-7-酮,具体结构式如下:
[0003][0004]
wo2007/056955公开了一类吡唑并嘧啶酮衍生物,制法及用途。该类衍生物经药理试验证明具有较强的pde5抑制活性,且毒性较低,该类衍生物在临床上可以用来改善或治疗心脑血管系统,泌尿系统症状或疾病,尤其可以用来改善或治疗包括勃起功能障碍在内的症状或疾病,其中包括tpn729,它在体外酶抑制剂筛选试验中,表现了对ped5酶极高的活性和选择性。
[0005]
目前合成tpn729的制备需要经过一个关键中间体n-甲基-2-吡咯烷乙胺,如下所示的化合物i,包括其游离碱和盐的形式:
[0006][0007]
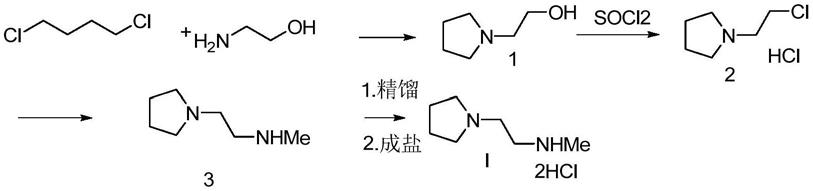
化合物i以往的合成路线如下:
[0008][0009]
该路线主要不足之处在于:(1)中间体1市场不易得,只能通过前期反应制备。(2)合成中间体2需要用到二氯亚砜,工业生产中污染大。(3)合成化合物3中甲氨取代同样副反应多,比如双取代生成杂质3-a,2自身及2和 3相互成季铵盐等,导致收率低。
[0010][0011]
鉴于上述以往的方法存在的许多不足,因此开发合成路线短,原子经济性高,生产成本低,易于工业化生产的制备方法具有很大的实际意义。
技术实现要素:
[0012]
技术问题
[0013]
本发明所要解决的技术问题是克服已有技术的不足,提供一种反应条件温和,工艺简便,且适合工业化的制备n-甲基-2-吡咯烷乙胺的方法。
[0014]
因此,本发明的目的是提供一种产率高,副产物少,绿色经济等优点,适合于开发为工业化生产工艺的n-甲基-2-吡咯烷乙胺的制备方法。
[0015]
技术方案
[0016]
根据一个方面,本发明提供了一种制备如下所述的式i所示的化合物或其盐的方法,该方法包括以下步骤:
[0017][0018][0019]
c)使化合物iv与化合物v经取代反应生成化合物vi,
[0020]
d)使化合物vi脱boc得到化合物i,
[0021]
其中,boc为叔丁氧羰基,ms为甲磺酰基。
[0022]
有益效果
[0023]
本发明首先使用易得的化工品作为起始原料制备n-甲基-2-吡咯烷乙胺及其盐酸盐。其优势在于合成过程中副产物少,且易于纯化,同时总收率较高,且没有大量污染,适合于开发为工业化生产工艺,符合绿色经济原则。
具体实施方式
[0024]
为使本领域具有普通知识的人员可了解本发明的特点及效果,以下谨就说明书及申请专利范围中提及的术语及用语进行一般性的说明及定义。除非另有指明,否则文中使用的所有技术及科学上的字词,皆具有本领域技术人员对于本发明所了解的通常意义,当
有冲突情形时,应以本说明书的定义为准。
[0025]
在本文中,用语“包含”、“包括”、“具有”、“含有”或其他任何类似用语均属于开放性连接词(open-ended transitional phrase),其意欲涵盖非排他性的包括物。举例而言,含有复数要素的一组合物或制品并不仅限于本文所列出的这些要素而已,而是还可包括未明确列出但却是该组合物或制品通常固有的其他要素。除此之外,除非有相反的明确说明,否则用语“或”是指涵盖性的“或”,而不是指排他性的“或”。例如,以下任何一种情况均满足条件“a或b”:a为真(或存在)且b为伪(或不存在)、a为伪(或不存在)且b为真(或存在)、a和b均为真(或存在)。此外,在本文中,用语“包含”、“包括”、“具有”、“含有”的解读应视为已具体公开并同时涵盖“由
…
所组成”及“实质上由
…
所组成”等封闭式或半封闭式连接词。
[0026]
在本文中,所有以数值范围或百分比范围形式界定的特征或条件仅是为了简洁及方便。据此,数值范围或百分比范围的描述应视为已涵盖且具体公开所有可能的次级范围及范围内的个别数值,特别是整数数值。举例而言,“1至8”的范围描述应视为已经具体公开如1至7、2至8、2至6、3至6、 4至8、3至8等等所有次级范围,特别是由所有整数数值所界定的次级范围,且应视为已经具体公开范围内如1、2、3、4、5、6、7、8等个别数值。除非另有指明,否则前述解释方法适用于本发明全文的所有内容,不论范围广泛与否。
[0027]
若数量或其他数值或参数是以范围、较佳范围或一系列上限与下限表示,则其应理解成是本文已特定公开了由任一对该范围的上限或较佳值与该范围的下限或较佳值构成的所有范围,不论这些范围是否有分别公开。此外,本文中若提到数值的范围时,除非另有说明,否则该范围应包括其端点以及范围内的所有整数与分数。
[0028]
在本文中,在可实现发明目的的前提下,数值应理解成具有该数值有效位数的精确度。举例来说,数字40.0则应理解成涵盖从39.50至40.49的范围。
[0029]
在本文中,对于使用马库什群组(markush group)或选项式用语以描述本发明特征或实例的情形,本领域技术人员应了解马库什群组或选项列表内所有要素的次级群组或任何个别要素亦可用于描述本发明。举例而言,若x 描述成“选自于由x1、x2及x3所组成的群组”,亦表示已经完全描述出x 为x1的主张与x为x1及/或x2的主张。再者,对于使用马库什群组或选项式用语以描述本发明的特征或实例的情况,本领域技术人员应了解马库什群组或选项列表内所有要素的次级群组或个别要素的任何组合亦可用于描述本发明。据此,举例而言,若x描述成“选自于由x1、x2及x3所组成的群组”,且y描述成“选自于由y1、y2及y3所组成的群组”,则表示已经完全描述出x为x1或x2或x3而y为y1或y2或y3的主张。
[0030]
以下具体实施方式本质上仅是例示性,且并不欲限制本发明及其用途。此外,本文并不受前述现有技术或发明内容或以下具体实施方式或实施例中所描述的任何理论的限制。
[0031]
根据本公开的一个实施方式,其提供了一种制备如下所述的式i所示的化合物或其盐的方法,该方法包括以下步骤:
[0032]
[0033][0034]
c)使化合物iv与化合物v经取代反应生成化合物vi,
[0035]
d)使化合物vi脱boc以得到化合物i,
[0036]
其中,boc为叔丁氧羰基,ms为甲磺酰基。
[0037]
通过上述方法,使用易得的化工品作为起始原料制备n-甲基-2-吡咯烷乙胺及其盐酸盐。其优势在于合成过程中副产物少,且易于纯化,同时总收率较高,且没有大量污染,适合于开发为工业化生产工艺,符合绿色经济原则。
[0038]
根据本公开的一个实施方式,其中,在步骤c)中,化合物v与化合物 iv的投料摩尔比为1~3:1,优选为1~1.3:1,更优选为1.05~1.1:1。在上述投料比下,可以促进反应向期望的方向顺利进行,提高转化率,并减少副产物的产生。
[0039]
根据本公开的一个实施方式,其中,步骤c)的反应温度为30℃至70℃,优选45℃至55℃,反应时间为8小时~24小时,优选为10~12小时。在上述反应温度和反应时间下,可以提高反应速度,提高转化率,并减少副产物的产生。
[0040]
根据本公开的一个实施方式,步骤d)的脱boc可以在任何适合的条件下进行,而没有特别限制,只要能够实现脱boc的目的且对于反应产物没有显著不利影响即可。例如,脱boc可以在酸存在下进行。所述酸可以是有机酸或者无机酸。所述的有机酸可以选自甲酸、乙酸、丙酸、草酸、马来酸、枸橼酸、酒石酸、甲磺酸、和苯磺酸;所述无机酸可以选自盐酸、硫酸和磷酸。特别地,所述酸是盐酸,此时,步骤d)可以得到式i化合物的盐酸盐。在使用上述的特定有机酸和无机酸的情况下,可以提高选择性,减少其他副产物的产生。
[0041]
在脱boc在酸存在下进行的情况下,可以得到式i化合物与该酸的盐。
[0042]
在脱boc在酸存在下进行的情况下,使用的酸与式vi化合物的投料摩尔比为2~3:1,优选为2.3~2.7:1,更优选为2.4~2.6:1。在上述投料比下,可以促进反应向期望的方向顺利进行,提高转化率,并减少副产物的产生。
[0043]
根据本公开的一个实施方式,其中,步骤d)的反应温度为-15℃至15℃,优选-10℃至10℃;反应时间为1小时~2小时。在上述反应温度和反应时间下,可以提高反应速度,提高转化率,并减少副产物的产生。
[0044]
根据本公开的另一个实施方式,其中,该方法在步骤c)之前进一步包括:
[0045]
a)使化合物ii与boc酸酐反应以生成化合物iii,
[0046]
b)使化合物iii与甲磺酰氯反应以生成化合物iv,
[0047]
即,该方法总的反应式如下:
[0048][0049]
其中,步骤b)可以在碱性试剂的作用下进行。所述碱性试剂可以为选自叔丁醇钾、三乙胺、二异丙基胺、二异丙基乙基胺、三正丁胺、吡啶的一种或几种的混合物。
[0050]
通过使用化合物ii作为起始原料,引入保护基后中间体易于后处理操作,最后脱保护基同时成盐,4步反应的总收率60%。其优势在于合成过程中副产物少,且易于纯化,同时四步反应总收率较高,且没有大量污染,适合于开发为工业化生产工艺,符合绿色经济原则。
[0051]
根据本公开的一个实施方式,其中,在步骤a)中,所述boc酸酐与式 ii化合物的投料摩尔比为1~1.05:1,优选为1~1.03:1,更优选为1~1.01:1。在上述投料比下,可以促进反应向期望的方向顺利进行,提高转化率,并减少副产物的产生。
[0052]
根据本公开的一个实施方式,其中,在步骤a)中,反应温度优选为-15℃至15℃,更优选为-10℃至10℃;反应时间为0.5小时~1小时。
[0053]
根据本公开的一个实施方式,其中,在步骤b)中,甲磺酰氯与化合物 iii的投料摩尔比为1~1.05:1,优选为1~1.03:1,更优选为1~1.01:1。在上述投料比下,可以促进反应向期望的方向顺利进行,提高转化率,并减少副产物的产生。
[0054]
根据本公开的一个实施方式,其中,在步骤b)中,所述的碱性试剂与化合物iii的投料摩尔比为1~1.05:1,优选为1~1.03:1,更优选为1~1.02:1。在上述投料比下,可以促进反应向期望的方向顺利进行,提高转化率,并减少副产物的产生。
[0055]
根据本公开的一个实施方式,其中,在步骤b)中,反应温度为-15℃至 15℃,优选-10℃至10℃;反应时间为0.5小时~2小时。在上述反应温度和反应时间下,可以提高反应速度,提高转化率,并减少副产物的产生。
[0056]
根据本公开的一个实施方式,其中,上述步骤a)-d)使用一锅法进行,而且,一锅法中使用的溶剂为选自烷烃、芳烃、卤代烃、酯类、醚类、极性非质子溶剂的一种或其两种以上的混合物,优选为二氯甲烷,甲苯。
[0057]
根据本公开的一个实施方式,其中,所述的溶剂与式ii化合物的质量体积比为5~12:1,优选为7~11:1,更优选为8~10:1。
[0058]
在使用一锅法,并使用上述特定的溶剂、以及溶剂含量的情况下,可以顺利地进行一锅法反应,减少中间的提纯步骤等的消耗。
[0059]
现通过以下的实施例来进一步描述本发明的有益效果,应理解为这些实施例仅用于例证的目的,不限制本发明的范围,同时本领域普通技术人员根据本发明所做的显而易见的改变和修饰也包含在本发明范围之内。
[0060]
实施例1化合物iii的合成
[0061][0062]
将化合物ii溶于甲苯溶剂中,冰水浴冷却至0-10℃,滴加boc2o,滴加过程中会放出气体,控制温度低于20℃,滴完后在0-10℃左右搅拌30min, tlc显示反应完后直接进行下一步;1h nmr(400mhz,氯仿-d)δ3.74(t,j= 5.4hz,2h),3.39(t,j=5.4hz,2h),2.92(s,3h),2.66(s,1h),1.46(s,9h).
13
c nmr(101mhz,氯仿-d)δ157.04,79.79,60.75,51.17,35.34,28.33.
[0063]
实施例2化合物iv的合成
[0064][0065]
将上述反应液冷却至0-10℃,加入三乙胺,滴加mscl,滴加过程中控制温度小于20℃,析出白色固体,滴完后搅拌30min。tlc显示反应完,将反应液过滤,滤饼用甲苯洗涤,滤液直接进行下一步;
[0066]
实施例3化合物vi的合成
[0067][0068]
将上述滤液氮气置换三次,控制温度低于20℃,滴加吡咯烷,滴完后温度将内温升至35-45℃搅拌16h以上,tlc显示原料iv反应完,后处理:将反应液降至20℃左右,加入水,搅拌分层,有机层再用水洗涤2次,有机层浓缩除去水分后,过滤,滤液浓缩干得到浅色油状物vi,质量收率: 170-250%.1h nmr(400mhz,氯仿-d)δ3.43
–
3.31(m,2h),2.88(s,3h),2.60 (d,j=10.7hz,6h),1.78(m,j=3.1hz,4h),1.46(s,9h).
13
c nmr(101mhz, 氯仿-d)δ155.57,79.16,54.20,53.85,53.44,48.07,47.65,34.51,28.36,23.37
[0069]
实施例4化合物i的合成
[0070][0071]
将vi降温至0-10℃,滴加浓盐酸,控制温度小于40℃,滴加过程放出气体,滴完后升温至55-65℃反应1h,tlc监控vi反应完全,浓缩除去水分,加异丙醇分散,再浓缩除去溶剂,再加异丙醇分散,浓缩除去溶剂,加 5倍体积异丙醇,加热回流溶清,后降温析出固体,在20℃左右搅拌3-4h,过滤,滤饼用异丙醇洗涤,60℃烘干得到类白色固体i,质量收率:60-85%。1h nmr(400mhz,氯仿-d)δ3.53
–
3.31(dt,j=79.2,6.8hz 8h),3.07(s,1h), 2.56(s,3h),1.93(s,4h).
13
c nmr(101mhz,dmso-d6)δ53.54,49.47,43.95, 32.65,23.13.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1