基于肠道微生物相对丰度的胆管癌非侵入性标志物、筛选方法及应用与流程
1.本发明涉及基因工程技术领域,特别是一种基于肠道微生物相对丰度的胆管癌非侵入性标志物、筛选方法及应用。
背景技术:
2.目前,胆管癌占原发性肝癌的10%
‑
20%,是第二常见的恶性肝肿瘤,近年来在全世界范围内发病率呈上升趋势。胆管癌是最具侵袭性和破坏性的恶性亚型之一,具有发病隐匿、生长快速、易通过淋巴和血液循环早期转移,预后不良的特点,晚期胆管癌患者存活率低于5%。胆管癌给公众健康带来了沉重的负担,是一个严重的全球性公众健康问题。
3.迄今为止,胆管癌被认为可能与遗传和环境因素有关,其特定的病因学仍然无法解释。研究发现,人体肠道内寄居着种类繁多的微生物,这些微生物称为肠道菌群,他们能影响消化能力、抵御感染和患病风险。肠道菌群携带约2.5万个基因,是人自身基因数的150倍,是疾病发生过程中的一个关键的环境因素。研究发现,不同疾病患者的肠道微生物群的组成不同,肠道微生物群与许多癌症的发展和进展有关。过去十年积累的证据表明,肠道菌群通过肠肝轴与肝脏疾病的发展关联,在肝癌的发展中起着至关重要的作用。由肠道微生物群变化引起的炎症信号已被认为是一种新的潜在致癌机制。这些研究证明了肠道菌群的诊断潜力。另外,关于胆管癌患者的肠道菌群特征的文献相对较少,我们尚无法将胆管癌患者与其他肝病患者的肠道菌群区分开来,这表明了进行胆管癌患者肠道菌群研究的必要性。
4.目前诊断胆管癌的金标准为手术切除的肿瘤组织的病理报告,胆管癌诊断的血清肿瘤标志物为ca19
‑
9,术前诊断准确率不高,仅有0.693。临床上缺乏针对胆管癌的有效非侵入性诊断标志物。
技术实现要素:
5.为了克服现有技术的不足,本发明提供了一种肠道微生物属标志物可以用于早期诊断胆管癌,通过对受试者的粪便进行16sr rna基因测序,测定粪便中特定肠道菌群属的丰度,判断受试者是否患有胆管癌,从而实现通过无创手段诊断胆管癌,并提高胆管癌患者的早期诊断准确率。
6.为了实现上述目的,本发明采用的技术方案是:一种基于肠道微生物相对丰度的胆管癌非侵入性标志物,包括12个肠道菌群属。
7.作为本发明的进一步设置,所述12个肠道菌群属及相应的系
8.数如下表:
9.genuscoefg__agathobacter0.114687157g__anaerostipes1.785618953
g__cetobacterium
‑
0.230269885g__clostridium
‑
0.320320014g__clostridium_sensu_stricto_10.104688944g__coprococcus_30.051689828g__faecalibacterium0.106173431g__intestinimonas
‑
1.399324406g__muribaculaceae_unclassified
‑
0.034562105g__muribaculum
‑
0.254203749g__rothia0.220752823g__turicibacter
‑
0.065048068
10.一种基于肠道微生物相对丰度的胆管癌非侵入性标志物的筛选方法,包括:
11.(1)收集受试者的粪便样本,受试者包括胆管癌患者和非癌症者,对粪便样本进行16s rrna基因检测;(2)dna提取和16s rrna基因测序:用e.z.n.a.stooldnakit(d4015,omega,inc.,usa)提取细菌基因组,对原核(细菌和古细菌)小亚基(16s)rrna基因的v3
‑
v4区用扩增,在illumina novaseq平台进行测序;
12.(3)生物信息学分析,使用qiime2软件包对原始读取数据进行分析,根据fqtrim(v0.94),在特定的过滤条件下对raw data进行质量过滤,以获得clean data,以100%相似性对clean data进行聚类得到feature(特征),dada2软件用于过滤测序读数并构建特征表和特征序列,物种注释的序列比对由blast完成,比对数据库为silva和nt
‑
16s;
13.(4)数据分析,利用α多样性,β多样性(pcoa)评估样本的总体差异;
14.(5)用wilcoxon秩和检验筛选两组间显著丰度差异的微生物共98个,然后用线性判别分析效应大小(lefse)分析再次识别胆管癌和非癌组之间的差异微生物共60个,将以上两组差异微生物交叉,可得到32个共同差异微生物;接着,利用lasso
‑
logistic回归(r包(v3.5.2))分析建立12个微生物属模型;
15.(6)用proc包建立模型的受试者roc曲线,分别计算每个属和12属组合的auc,以评价其作为标记物的性能,并在muribaculaceae_unclassified属中得到了较高的auc=0.84,12个属的组合作为预测因子得到的auc=0.93。
16.一种基于肠道微生物相对丰度的胆管癌非侵入性标志物的应用,所述胆管癌的测定方法如下:
17.(1)采集受检者的粪便标本进行16s rrna基因测序,获取12个肠道菌群属的相对丰度;
18.(2)将12个肠道菌群属的相对丰度与各自相应的系数相乘,并将相乘后的各个结果相加,得到riskscore,当riskscore超过cutoff值即可判断受检者患有胆管癌,cutoff值为0.590917897。
19.结合上述的所有技术方案,本发明所具备的优点及积极效果为:通过对受试者的粪便进行16sr rna基因测序,测定粪便中特定肠道微生物属的丰度,判断受试者是否患有胆管癌,从而实现通过无创手段诊断胆管癌,并提高胆管癌患者的早期诊断准确率。
20.下面结合附图对本发明作进一步描述。
附图说明
21.附图1为胆管癌和非癌中观测的feature数和simpson多样性指数差异图;
22.附图2为基于非加权(unifrac distance metric)的pcoa图;
23.附图3为lefse分析图;
24.附图4为使用lasso
‑
logistic建立基于肠道菌群特征的疾病诊断模型图;
25.附图5为使用roc曲线评价12个微生物属单独及其组合的胆管癌诊断效果图;
26.附图6为使用随机森林法比较12个微生物属组合和临床变量对cca的影响图。
具体实施方式
27.本发明的具体实施例如图1
‑
6所示,收集胆管癌患者33名和非癌症47名受试者的粪便样本,对粪便样本进行16s rrna基因检测。调查记录人口统计学基线数据,包括年龄、性别、体重、身高、吸烟和饮酒习惯等,追踪血清肿瘤标记物、肝功能指标。临床变量中,性别、吸烟和饮酒习惯、肝硬化和hbv感染史,在cca和cf之间具有可比性。
28.dna提取和16s rrna基因测序:
29.首先,用e.z.n.a.stooldnakit(d4015,omega,inc.,usa)提取细菌基因组。对原核(细菌和古细菌)小亚基(16s)rrna基因的v3
‑
v4区用扩增。在整个脱氧核糖核酸提取过程中,超纯水被用来排除作为阴性对照的假阳性聚合酶链反应结果的可能性。聚合酶链反应产物用ampure xt beads(beckman coulter genomics,danvers,ma,usa)纯化,用qubit(invitrogen,usa)定量。扩增子池用于测序。agilent 2100生物分析仪(agilent,usa)和illumina的文库定量试剂盒(美国马萨诸塞州沃本市卡帕生物科学公司)用于评估扩增子文库的大小和数量。样品在illumina novaseq平台上进行测序。
30.生物信息学分析:
31.根据fqtrim(v0.94),在特定的过滤条件下对原始读数进行质量过滤,以获得高质量的干净标签。以100%相似性进行聚类得到feature(特征)。dada2软件用于过滤测序读数并构建特征表和特征序列。利用mafft软件(v 7.310)对不同类群优势种的差异进行多重序列比对。物种注释的序列比对由blast完成,比对数据库为silva和nt
‑
16s。
32.数据分析:
33.利用chao1、observing species,goods_coverage,shannon和simpson描述样本的α多样性。β多样性(pcoa)用于评估样本的总体差异。然后,用非参数检验筛选两组间显著丰度差异的微生物共98个,然后用线性判别分析效应大小(lefse)分析再次识别胆管癌和非癌组之间的差异微生物共60个。将以上两组差异微生物交叉,我们得到了32个共同差异微生物。接着,利用lasso
‑
logistic回归分析确定了12个最能区分胆管癌和非癌的属。我们分别用单个属和12个属组合作为预测因子,生成了各自的auc,并在muribaculaceae_unclassified属中得到了较高的auc,为0.84,12个属的组合作为预测因子得到的auc为0.93,能最好地区分胆管癌和非癌。最后应用随机森林算法消除临床变量对微生物的影响。
34.胆管癌和非癌组的alpha
‑
diversity和beta
‑
diversity比较,两组之间差异显著。图1胆管癌和非癌中观测的feature数和simpson多样性指数差异。箱型图表示四分位数(iqr),箱型图中的中线表示中位数。图2基于非加权(unifrac distance metric)的pcoa展示了胆管癌与非癌组的菌群组成差异显著(p=0.032)。具体表现为,有3个门、79个属及204
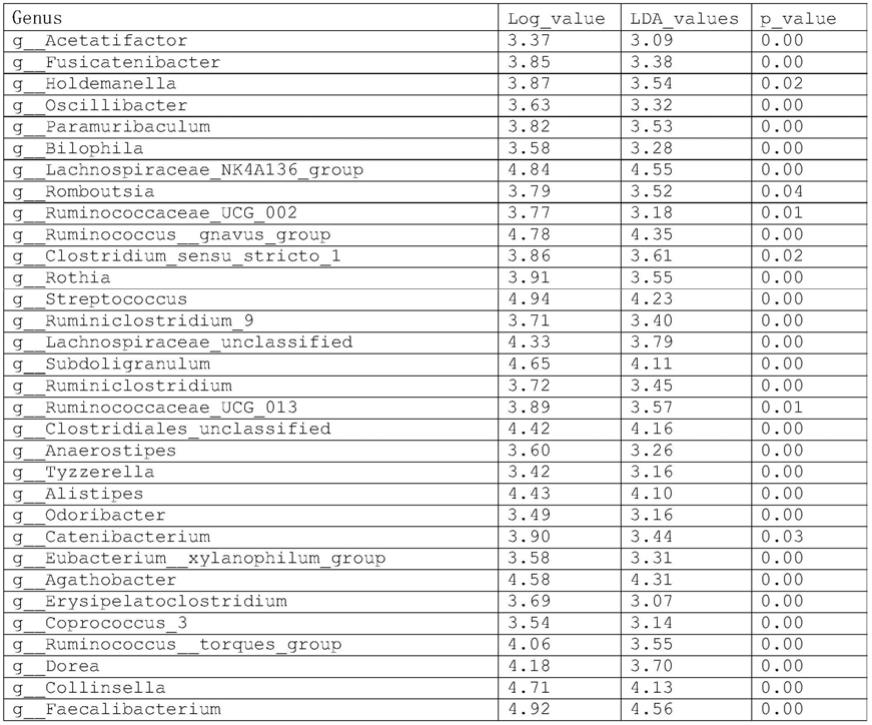
个种仅存在于胆管癌队列中,而5个门、106个属及248个种仅存在于非癌队列中。图3展示了两组之间9个显著差异门。图3lefse分析显示,两组之间有60个显著差异的微生物属。(lda评分>3.0,p<0.05)。具体微生物名称如下表1:
35.[0036][0037]
表2:胆管癌和非癌组之间显著差异细菌。使用wilcoxon秩和检验,筛选得到98个差异细菌属。
[0038]
[0039]
[0040]
[0041][0042]
表3:根据非参数检验和lefse,找到32个共同差异表达微生物属。
[0043]
[0044][0045]
如图4所示,建立基于肠道菌群特征的疾病诊断模型,使用lasso
‑
logistic建立了包含12个肠道微生物属的模型。12个微生物模型具体内容及系数见下表4:
[0046][0047][0048]
胆管癌的测定方法如下:
[0049]
(1)采集受检者的粪便标本进行16s rrna基因测序,获取12个肠道菌群属的相对丰度;
[0050]
(2)将12个肠道菌群属的相对丰度与各自相应的系数相乘,并将相乘后的各个结果相加,得到riskscore,当riskscore超过cutoff值即可判断受检者患有胆管癌,cutoff值为0.590917897。
[0051]
如图5所示,使用roc曲线评价12个微生物属单独及其组合的胆管癌诊断效果。
[0052]
如图6所示,使用随机森林法比较12个微生物属组合和临床变量的重要性,结果显示,12个属的组合对cca的影响大于所有临床变量。
[0053]
本发明不局限于上述具体实施方式,本领域一般技术人员根据本发明公开的内容,可以采用其他多种具体实施方式实施本发明的,或者凡是采用本发明的设计结构和思路,做简单变化或更改的,都落入本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1