一种化合物2,4,6-三氟苄胺的制备方法与流程
1.本发明涉及一种农药、医药中间体2,4,6-三氟苄胺的制备方法,属于化工化学技术领域,具体涉及化学合成领域。
背景技术:
2.2,4,6-三氟苄胺是一种重要的农药、医药中间体,在药物合成等领域具有广泛的用途,也是合成药物bictegravir的关键中间体,具有较高的市场前景和价值。
3.目前已公开的2,4,6-三氟苄胺的合成方法主要有以下三种:
4.①
专利w09845268公开了以2-溴甲基-1,3,5-三氟苯为起始原料,与乌洛托品成盐,然后酸性条件下水解得到2,4,6-三氟苄胺。该方法原料2-溴甲基-1,3,5-三氟苯原料较难得到,且价格昂贵,缺乏工业化生产价值。
5.②
专利cn104610068公开了以1,3,5-三氟苯为起始原料,经正丁基锂锂化、甲酰化、还原、氯化和置换反应得到2,4,6-三氟苄胺。该方法合成步骤长,反应操作条件复杂,锂化需要在超低温下进行,同时三废产生量也大,不利于大规模工业生产。
6.③
专利cn107778183公开了以五氯苯腈为起始原料,经氟化和两步氢化还原得到2,4,6-三氟苄胺。该方法虽然反应步骤短,原料也便宜,但两步加氢反应所用催化剂钯碳价格昂贵且难回收套用,同时加氢反应副产物多,收率低,造成工业化生产效益不高。
技术实现要素:
7.针对现有技术存在的上述问题,本发明目的是在于提供一种反应步骤短、原料成本低、操作简便、反应收率高、适合大规模工业化生产的2,4,6-三氟苄胺的合成方法。
8.为了实现上述目的,本发明采用如下技术方案:
9.一种化合物2,4,6-三氟苄胺的制备方法包括如下步骤:
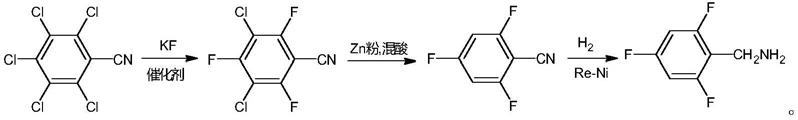
10.(a)以五氯苯腈为原料,在有机溶剂中和催化剂的作用下与氟化剂进行氟化反应得到3,5-二氯-2,4,6-三氟苯腈。
11.(b)将(a)中得到的3,5-二氯-2,4,6-三氟苯腈,加入水中,与锌粉和混酸反应,回流脱氯得到2,4,6-三氟苯腈。
12.(c)将(b)中得到的2,4,6-三氟苯腈,加入有机溶剂中,在有机碱和催化剂的作用下,加氢还原得到产品2,4,6-三氟苄胺。
13.作为优先方案,所述步骤(a)中所用有机溶剂为:环丁砜。溶剂用量与五氯苯腈的质量比为(3-6):1,所述氟化剂为氟化钾或氟化钠,氟化剂与五氯苯腈的摩尔比为(3.1-5):1,反应温度为110℃-150℃,反应时间为2h-12h。进一步更优先,氟化剂与五氯苯腈的摩尔比为(3.1-4.0):1,反应温度为125℃-135℃,反应时间为4h-6h。
14.作为优先方案,所述步骤(a)中所用催化剂为下列催化剂中的任意一种:四丁基溴化铵、四丁基氯化铵、18-冠醚-6等,所用催化剂与五氯苯腈的质量比为(0.02-0.1):1。进一步更优先,所用催化剂与五氯苯腈的质量比为(0.03-0.05):1。
15.作为优先方案,所述步骤(b)中溶剂水用量与3,5-二氯-2,4,6-三氟苯腈的质量比为(8-20):1,进一步更优先,溶剂水用量与3,5-二氯-2,4,6-三氟苯腈的质量比为(10-15):1,所用锌粉与3,5-二氯-2,4,6-三氟苯腈摩尔比为(2-5):1,回流反应温度为95℃-105℃,反应时间为3h-16h。
16.实验发现,用锌粉加醋酸、盐酸或硫酸可脱除3,5-二氯-2,4,6-三氟苯中的二个氯,而且惊奇在发现,用锌粉与单一酸脱氯的效率没有与混酸脱氯的效率高。脱氯反应在水溶液中进行,用锌粉与醋酸脱氯反应速度太慢,脱氯不完全,结果得到的2,4,6-三氟苯腈收率不高;用锌粉与盐酸或硫酸脱氯,锌粉消耗速度太快,副产物增多,有效反应的效率低,结果得到的2,4,6-三氟苯腈收率也不高;而用锌粉与混酸脱氯反应能得到效好的收率,这里的混酸是指醋酸与盐酸或硫酸的一定比例混合物。
17.作为优先方案,所述步骤(b)中,所述的混酸为浓盐酸或浓硫酸与醋酸的混合物,浓盐酸或浓硫酸与3,5-二氯-2,4,6-三氟苯腈的摩尔比为(2-5):1,所用醋酸与3,5-二氯-2,4,6-三氟苯腈的摩尔比为(5-20):1。进一步更优先,所用醋酸与3,5-二氯-2,4,6-三氟苯腈的摩尔比为(10-15):1。
18.作为优先方案特别说明,本发明所述步骤(b)脱氯反应使用混酸为浓盐酸或浓硫酸与醋酸的混合物,单一的使用某一种酸,会造成反应脱氯不完全,副产物增多,使得反应收率严重下降。所述步骤(b)中脱氯反应中,使用的混酸(浓盐酸或浓硫酸与醋酸)摩尔比为1:(2-7)。
19.作为优先方案特别说明,本发明所述步骤(b)脱氯反应使用的混酸加料方式采用缓慢滴加方式,防止反应速度过快。
20.作为优先方案,所述步骤(c)中,有机溶剂为下列溶剂中的任意一种:甲苯、环己烷、正己烷、正庚烷等,所用溶剂量与2,4,6-三氟苯腈的质量比为(5-10):1,所述的有机碱为二异丙基乙基胺或三乙胺,所用有机碱与有机溶剂的质量比为(0.02-0.1):1。
21.作为优先方案,所述步骤(c)中,所述的加氢催化剂为海绵状雷尼镍,所用催化剂与2,4,6-三氟苯腈的质量比(0.02-0.1):1,加氢还原反应温度为90℃-150℃,氢气压力为0.5mpa-3.0mpa,反应时间为2h-12h。进一步更优先,加氢还原反应温度为110℃-130℃,氢气压力为1.0mpa-2.0mpa,反应时间为3h-6h。
22.作为优先方案,所述步骤(c)加氢反应要在碱性环境下进行,所述的碱优选为有机碱二异丙基乙基胺或三乙胺。我们实验发现,若加氢反应在酸性条件下进行,反应副产物多,收率低。
23.本发明的具体合成反应路线如下:
[0024][0025]
本发明有益效果如下:
[0026]
本发明合成方法反应步骤短、原料成本低、操作简便、反应收率高、适合大规模工业化生产的2,4,6-三氟苄胺。
[0027]
本发明步骤(b)中脱氯反应使用混酸为浓盐酸或浓硫酸与醋酸的混合物,研究发
现单一的使用某一种酸,会造成反应脱氯不完全,副产物增多,使得反应收率严重下降。所述步骤(b)中脱氯反应中,使用的混酸提高反应收率。
[0028]
本发明步骤(b)脱氯反应使用的混酸加料方式采用缓慢滴加方式,防止反应速度过快。
[0029]
实验发现加氢反应在酸性条件下进行,反应副产物多,收率低;因此本发明步骤(c)的加氢反应在碱性环境下进行。
具体实施方式
[0030]
下面结合实施例对本发明技术方案做进一步完整的说明
[0031]
实施例1:
[0032]
在2000ml三口瓶中,加入200g五氯苯腈(0.726mol)、160g无水氟化钾(2.75mol)、1000g环丁砜,搅拌,氮气保护下,再加入6.0g18-冠醚-6,升温至125℃-135℃回流反应4h。反应完全后,降温至50℃以下,过滤,滤液加入2000ml水、800ml环己烷,搅拌,分层,取有机层用400ml水洗,分层,用无水硫酸钠干燥,过滤,滤液减压蒸除溶剂,得到154.8g3,5-二氯-2,4,6-三氟苯腈(收率94.3%,hplc纯度98.7%)。
[0033]
实施例2:
[0034]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、23.2g锌粉(0.355mol),搅拌,升温至90℃,滴加43.1g30%浓盐酸(0.355mol)与80g醋酸(1.332mol)的混合物(混酸摩尔比1:3.8),滴加持续2h,然后在95℃-105℃保温回流4h,反应完全后,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得11.0g2,4,6-三氟苯腈(收率79.1%,gc纯度97.2%)。
[0035]
实施例3:
[0036]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、23.2g锌粉(0.355mol),搅拌,升温至90℃,滴加43.1g30%浓盐酸(0.355mol)与60g醋酸(0.999mol)的混合物(混酸摩尔比1:2.8),滴加持续2h,然后在95℃-105℃保温回流5h,反应完全后,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得10.7g2,4,6-三氟苯腈(收率77.0%,gc纯度97.5%)。
[0037]
实施例4:
[0038]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、17.4g锌粉(0.265mol),搅拌,升温至90℃,滴加43.1g30%浓盐酸(0.355mol)与60g醋酸(0.999mol)的混合物(混酸摩尔比1:2.8),滴加持续2h,然后在95℃-105℃保温回流6h,反应完全后,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得10.1g2,4,6-三氟苯腈(收率72.7%,gc纯度96.5%)。
[0039]
实施例5:
[0040]
在500ml20g三口瓶中加入3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、29g锌粉(0.4434mol),搅拌,升温至90℃,滴加43.1g30%浓盐酸(0.355mol)与80g醋酸
(1.332mol)的混合物(混酸摩尔比1:3.8),滴加持续2.0h,然后在95℃-105℃保温回流4h,反应完全后,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得11.1g2,4,6-三氟苯腈(收率79.8%,gc纯度97.1%)。
[0041]
实施例6:
[0042]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、23.2g锌粉(0.355mol),搅拌,升温至90℃,滴加30%浓盐酸32.3g(0.266mol)与醋酸80g(1.332mol)的混合物(混酸摩尔比1:5),滴加持续2h,然后在95℃-105℃保温回流5h,反应完全后,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得10.9g2,4,6-三氟苯腈(收率78.4%,gc纯度97.1%)。
[0043]
实施例7:
[0044]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、(23.2g锌粉0.3547mol),搅拌,升温至90℃,滴加26.9g30%浓盐酸(0.221mol)与80g醋酸(1.332mol)混合物(混酸摩尔比1:6),滴加持续2h,然后在95℃-105℃保温回流5h,反应完全后,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得10.3g2,4,6-三氟苯腈(收率74.1%,gc纯度96.9%)。
[0045]
实施例8:
[0046]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、23.2g锌粉(0.355mol),搅拌,升温至90℃,滴加26.6g浓硫酸(0.271mol)与80g醋酸(1.332mol)的混合物(混酸摩尔比1:4.9),滴加持续2h,然后在95℃-105℃保温回流5h,反应完全后,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得10.4g2,4,6-三氟苯腈(收率74.8%,gc纯度96.8%)。
[0047]
对比实施例1:
[0048]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、23.2g锌粉(0.355mol),搅拌,升温至90℃,滴加醋酸80g(1.332mol),滴加持续2h,然后在95℃-105℃保温回流16h,(gc检测脱氯不完全)降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得2.7g2,4,6-三氟苯腈(收率19.4%,gc纯度95.7%)。
[0049]
对比实施例2:
[0050]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、23.2g锌粉(0.355mol),搅拌,升温至90℃,滴加32.3g30%浓盐酸(0.266mol),滴加持续2h,然后在95℃-100℃保温回流6h,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得8.7g2,4,6-三氟苯腈(gc纯度66.7%)。
[0051]
对比实施例3:
[0052]
在500ml三口瓶中加入20g3,5-二氯-2,4,6-三氟苯腈(0.0885mol)、300g水、23.2g
锌粉(0.355mol),搅拌,升温至90℃,滴加26.6g浓硫酸(0.271mol),滴加持续2h,然后在95℃-100℃保温回流6h,降至室温,加入250ml二氯甲烷搅拌,过滤,滤液分层,取有机层用饱和碳酸氢钠水溶液调ph至7,再分层,有机层用150ml水洗后,无水硫酸镁干燥,过滤,滤液减压蒸馏得9.2g2,4,6-三氟苯腈(gc纯度60.9%)。
[0053]
实施例9:
[0054]
在500ml加氢釜中,加入10g2,4,6-三氟苯腈(0.0637mol)、0.3g雷尼镍、80g甲苯、4g三乙胺,分别用氮气、氢气置换釜内气体三次,然后通入氢气压力1.5mpa,控温130℃-140℃,搅拌反应5h。反应完全后,降至室温,过滤,回收雷尼镍,滤液减压蒸馏得到8.6g2,4,6-三氟苄胺(收率83.9%,gc纯度99.4%)。
[0055]
实施例10:
[0056]
在500ml加氢釜中,加入10g2,4,6-三氟苯腈(0.0637mol)、0.4g雷尼镍、100g甲苯、5g三乙胺,分别用氮气、氢气置换釜内气体三次,然后通入氢气压力2.0mpa,控温115℃-125℃,搅拌反应4h。反应完全后,降至室温,过滤,回收雷尼镍,滤液减压蒸馏得到8.9g2,4,6-三氟苄胺(收率86.8%,gc含量99.3%)。
[0057]
实施例11:
[0058]
在500ml加氢釜中,加入10g2,4,6-三氟苯腈(0.0637mol)、0.3g雷尼镍、80g环己烷、4.8g二异丙基乙基胺,分别用氮气、氢气置换釜内气体三次,然后通入氢气压力1.5mpa,控温120℃-130℃,搅拌反应5h。反应完全后,降至室温,过滤,回收雷尼镍,滤液减压蒸馏得到8.5g2,4,6-三氟苄胺(收率82.9%,gc含量99.6%)。
[0059]
对比实施例4:
[0060]
在500ml加氢釜中,加入10g2,4,6-三氟苯腈(0.0637mol)、1.0g10%钯碳、100g四氢呋喃、12.5g硫酸(0.128mol),分别用氮气、氢气置换釜内气体三次,然后通入氢气压力1.5mpa,控温80℃左右,搅拌反应12h。反应完全后,降至室温,过滤,滤液减压蒸除溶剂,残余物加入150ml二氯甲烷,0℃-10℃用液碱调ph值至10左右,分层,有机层干燥,过滤,滤液减压蒸馏得到4.2g2,4,6-三氟苄胺(收率41.0%,gc含量99.1%)。
[0061]
以上所述仅为本发明的优选实施例而已,不能被认为用于限定本发明的实施范围,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1