一种脯氨酸恒格列净的合成方法与流程
[0001]
本发明涉及药物化学领域,具体涉及一种脯氨酸恒格列净的合成方法。
背景技术:
[0002]
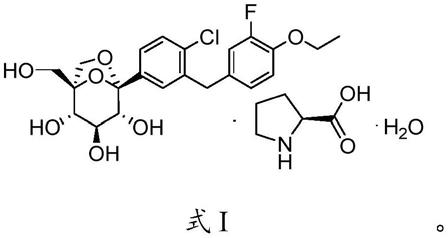
脯氨酸恒格列净是由江苏恒瑞医药股份有限公司自主研发的降血糖药。脯氨酸恒格列净可抑制sglt2(钠-葡萄糖转运蛋白2)对葡萄糖重吸收,增加尿中的排糖量,从而调节体内的血糖平衡。脯氨酸恒格列净的化学名称为:1,6-脱水-1-c-{4-氯-3-[(3-氟-4-乙氧苯基)甲基]苯基}-5-c-(羟甲基)-β-l-艾杜吡喃糖l-脯氨酸一水合物,其化学结构如式i所示
[0003][0004]
专利wo2012019496公开了脯氨酸恒格列净的合成方法,该方法从三甲基硅烷基保护的葡萄糖内酯出发,在丁基锂的作用下与4-[(5-溴-2-氯苯基)甲基]-1-乙氧基-2-氟苯缩合,所得缩合物经叔丁基二甲基氯硅烷选择性保护糖的伯羟基再经苄基保护糖的仲羟基,然后将全保护的糖在弱酸性条件下脱去叔丁基二甲基硅保护基,所得伯醇经swern氧化得到醛糖,而后醛糖与多聚甲醛在强碱性条件下发生歧化反应获得二醇,随后二醇在酸性条件下分子内醚化并经还原氢化获得恒格列净游离物,最后该游离物与脯氨酸在含水溶剂中形成脯氨酸恒格列净一水合物,其合成路线如下所示:
[0005][0006]
现有技术中恒格列净合成工艺需通过保护基的变换策略,选择性保护糖羟基获得所需醛糖,保护脱保护步骤导致合成步骤冗长,中间体多数为粘稠液体,纯化困难,工业生产需多步柱层析,导致生产成本增加,不利于工业化生产。因此,需开发步骤简捷的易于纯化的低成本工业化路线。
技术实现要素:
[0007]
为了克服现有技术的不足,本发明的目的是提供一种脯氨酸恒格列净的合成方法,该方法操作步骤简捷,产物易于纯化,成本低,适合工业化。
[0008]
本发明的脯氨酸恒格列净的合成方法以三甲基硅烷基(tms)保护的d-葡萄糖内酯化合物2为起始原料,与溴代芳烃化合物3在正丁基锂的作用下缩合后直接在甲磺酸(msoh)作用下脱三甲基硅烷基保护获得芳基糖化合物4,在咪唑的存在下芳基糖化合物4与过量三甲基氯硅烷(tmscl)将糖的四个羟基全部保护后直接在对甲苯磺酸吡啶盐(ppts)作用下选择性脱去伯醇上的三甲基硅烷基保护获得伯醇化合物5,伯醇化合物5被三氧化硫吡啶氧化得到醛化合物6,随后醛化合物6在强碱性条件下与多聚甲醛发生歧化反应获得二醇化合物
7,二醇化合物7在酸性条件下分子内醚化获得恒格列净游离物化合物8,最后恒格列净游离物化合物8与脯氨酸在含水溶剂中形成脯氨酸恒格列净一水合物化合物1,其合成路线如下所示:
[0009][0010]
具体地,本发明的脯氨酸恒格列净的合成方法包括以下步骤:
[0011]
步骤1:化合物2与化合物3在正丁基锂的作用下缩合后直接在甲磺酸(msoh)作用下脱三甲基硅烷基保护获得化合物4;
[0012]
其中化合物3与正丁基锂的摩尔比为1:1.5~2.0,优选1:1.6;
[0013]
化合物3与化合物2的摩尔比为1:1~1.5,优选1:1.1;
[0014]
化合物3与甲磺酸的摩尔比为1:2.0~2.5,优选1:2.4;
[0015]
步骤1中的子步骤1)的反应温度为-90℃~-70℃,优选-85℃~-75℃,步骤1中的子步骤2)的反应温度为20℃~35℃,优选25℃~30℃;
[0016]
步骤2:在有机溶剂中,在咪唑的存在下,化合物4与三甲基氯硅烷(tmscl)反应将化合物4的四个羟基全部保护后直接在对甲苯磺酸吡啶盐(ppts)作用下选择性脱去伯醇上的三甲基硅烷基保护获得化合物5;
[0017]
其中化合物4和三甲基氯硅烷的摩尔比为1:5.0-6.0,优选的是1:5.5;
[0018]
化合物4和咪唑的摩尔比为1:5.5-6.5,优选的是1:6.0;
[0019]
化合物4和对甲苯磺酸吡啶盐的摩尔比为1:2.0-3.0,优选的是1:2.5;
[0020]
步骤2中的子步骤1)的反应温度为15℃~30℃,优选20℃~25℃,步骤2中的子步
骤2)的反应温度为5℃~20℃,优选10℃~15℃;
[0021]
有机溶剂选自二氯甲烷,甲苯,四氢呋喃等,优选的是二氯甲烷;
[0022]
步骤3:在有机溶剂中,在碱的存在下,化合物5被三氧化硫吡啶氧化得到化合物6;
[0023]
其中碱选自三乙胺,二异丙基乙胺等三级脂肪胺;
[0024]
化合物5与碱和三氧化硫吡啶的摩尔比为1:3.5~4.0:2.5~3.0,优选的是1:3.6:2.8;
[0025]
反应温度为-5℃~15℃,优选0℃~10℃;
[0026]
有机溶剂选自二氯甲烷,甲苯,四氢呋喃等,优选的是二氯甲烷;
[0027]
步骤4:在有机溶剂中,在碱的存在下,化合物6与多聚甲醛发生歧化反应获得化合物7;
[0028]
其中碱选自乙醇钠、氢氧化钾等;
[0029]
化合物6和多聚甲醛、碱的摩尔比为1:18~30:1.5~2.5,优选的是1:20:2.0;
[0030]
有机溶剂选自甲醇、乙醇、正丙醇、异丙醇等,优选乙醇;
[0031]
步骤5:在酸的存在下,化合物7分子内醚化获得化合物8;
[0032]
其中酸选自盐酸、硫酸或磷酸,优选盐酸,更优选30%盐酸;
[0033]
化合物7和酸的质量比(g/g)为1:0.2~1:0.5,优选1:0.3;
[0034]
反应温度为-5℃~15℃,优选0℃~10℃;
[0035]
步骤6:化合物8与脯氨酸在含水溶剂中形成化合物1;
[0036]
其中化合物8与脯氨酸的摩尔比为1:1.0~1.2,优选的是1:1.1;
[0037]
含水溶剂选自含水乙醇,优选95%乙醇(含水5%);
[0038]
反应温度为40℃~70℃,优选50~60℃。
[0039]
与现有技术相比,本发明的优势在于:
[0040]
(1)本发明合成方法步骤简捷,通过选择合适的羟基保护基有效克服了现有技术中保护基的变换带来的步骤冗长的弊端。
[0041]
(2)本发明各步反应收率较高,关键中间体易于分离提纯,有效降低生产的成本,适合工业化生产。
具体实施方案
[0042]
下列实施例进一步解释说明本发明,但是,它们并不构成对本发明范围的限制或限定。
[0043]
本发明实施例中使用的试剂均为市售常规试剂。
[0044]
实施例1化合物4的制备
[0045][0046]
氮气保护下,向1l四口瓶中投入60.0g 4-溴-1-氯-2-(4-乙氧基-3-氟苯甲基)苯
化合物3、108.0g thf、312g甲苯;降温至-85℃~-75℃,控温-85℃~-75℃滴加正丁基锂正己烷溶液(280ml,1mol/l),滴加时间1~2小时,滴毕,保温2小时;控温-85℃~-75℃滴加90.0g化合物2的甲苯(78.0g)溶液,滴加时间1~2小时,滴毕,控温-85℃~-75℃保温2小时;控温-50℃以下滴加40.5g甲磺酸的甲醇(120.0g)溶液,滴加时间1~2小时,滴毕;升温至25℃~30℃保温反应20小时。反应结束后将合成液降温至5℃~15℃,滴加配制好的10%naoh水溶液(10g naoh+90.0g水)调ph值至7~8,静置30分钟,分去下层水层。有机层减压脱溶,残余物加入120.0g甲醇,120g正己烷搅拌15分钟,静置,将上层(正己烷层)弃去;甲醇层浓缩至干,残余物加入120g甲醇34g甲苯,升温至40~50℃保温30分钟后,加入晶种控制温度-15℃~-10℃析晶,将所得晶体过滤,真空干燥得52g化合物4,收率为65%。esi-hrms(m/z):c
22
h
27
clfo7[m+h
+
]理论计算值:457.1424,实测值:457.1419。
[0047]
实施例2化合物5的制备
[0048][0049]
氮气保护下,向1l四口瓶中投入30.0g化合物4、26.8g咪唑、400g二氯甲烷,降温至0℃~10℃,控温0℃~10℃滴加39.2g三甲基氯硅烷,滴加时间2~3小时,滴毕,缓慢升温至20℃~25℃,保温2小时,反应结束,控制温度20℃以下,滴加150g水,保温搅拌20分钟,静置分层,有机层加入咪唑水溶液(0.1g咪唑加150g水)洗涤3次,而后氮气保护下,将二氯甲烷层降温至10℃~15℃,控制内温10℃~15℃,滴加配制好的4-甲基苯磺酸吡啶(对甲苯磺酸吡啶盐)水溶液(42.9g 4-甲基苯磺酸吡啶+43.0g水),控温10℃~15℃反应10小时,反应结束,分去水层,有机层加入3.2g磷酸二氢钠,14g磷酸氢二钠,200g水配制成的水溶液,静置分层,有机层用30.0元明粉,搅拌30分钟,过滤,滤液减压脱溶至干得42g粘稠液体化合物5,收率为95%。esi-hrms(m/z):c
31
h
51
clfo7si3[m+h
+
]理论计算值:673.2610,实测值:673.2618;1h-nmr(400mhz,cd3od)δ:7.26(s,1h),7.18(m,2h),6.72(m,3h),3.87-3.80(m,5h),3.61(m,1h),3.53(m,1h),3.39(m,1h),3.33(m,1h),3.09(m,1h),2.84(s,3h),1.15(t,j=6.4hz,3h),0.00(s,9h),,-0.02(s,9h),-0.58(s,9h);
13
c-nmr(100mhz,cdcl3)δ:153.34(d,j
c-f
=244hz),146.07(d,j
c-f
=11hz),138.62(d,j
c-f
=24hz),134.75,133.64,132.13,129.80,129.02,125.12,125.09,116.88(d,j
c-f
=19hz),115.63,102.85,79.10,75.80,,74.82,72.90,65.51,61.80,38.73,14.80,1.27,0.86,0.01。
[0050]
实施例3:化合物6的制备
[0051][0052]
氮气保护下,向四口瓶中投入30.0g化合物5,200g二氯甲烷,降温至0℃~10℃,控
温0℃~10℃,16.2g滴加三乙胺,继续滴加125.3g dmso,滴毕,加入400g二氯甲烷,分三次加入19.9g三氧化硫吡啶,控温0℃~10℃保温反应6小时,反应结束,控温15℃以下,滴加150g饮用水,搅拌后静置,分去上层水层。水层加入150g二氯甲烷,搅拌静置,分去上层水层。合并有机层,有机层用饱和氯化铵水溶液(氯化铵60g,水200g)洗涤后减压脱干,得42g油状物化合物6,收率为95%。esi-hrms(m/z):c
31
h
49
clfo7si3[m+h
+
]理论计算值:671.2453,实测值:671.2459。
[0053]
实施例4:化合物7的制备
[0054][0055]
在氮气保护下,向1l四口瓶中投入40g化合物6、400g乙醇、35.7g多聚甲醛,升温至50℃~55℃,滴加20%乙醇钠乙醇溶液39.0g,滴毕于50℃~55℃保温反应5小时。反应结束后控温50℃~55℃,滴加亚硫酸氢钠水溶液(亚硫酸氢钠112.0g,饮用水400g),控温45℃~55℃减压脱溶至无液体蒸出,而后加入300g甲基叔丁基醚,搅拌30分钟,静置,分去下层水层,将有机层控温40℃~45℃减压脱干,得26.1g白色固体化合物7,收率90%,esi-hrms(m/z):c
23
h
29
clfo8[m+h
+
]理论计算值:487.1530,实测值:487.1526。
[0056]
实施例5:化合物8的制备
[0057][0058]
氮气保护下,向250ml四口瓶中,加入10.0g化合物7,130g二氯甲烷,降温至-10℃~0℃,滴加30%盐酸3g,滴毕,缓慢升温至0℃~10℃保温反应22小时,反应完毕,控制0℃~10℃,滴加配制好的饱和碳酸氢钠水溶液,调ph值至7~8,残余物减压脱溶至干,加入50g乙酸乙酯,静置分层,有机层用200g水洗涤,而后减压脱干得8.8g化合物8,收率为95%,esi-hrms(m/z):c
22
h
25
clfo7[m+h
+
]理论计算值:455.1267,实测值:455.1260。1hnmr(400mhz,cd3od):δ7.49(s,1h),7.44-7.37(m,2h),6.97-6.89(m,3h),4.18-4.16(m,1h),4.08-4.04(m,4h),3.87-3.72(m,2h),3.69-3.56(m,4h),1.39(t,3h)。
[0059]
实施例6:脯氨酸恒格列净化合物1的制备
[0060]
[0061]
氮气保护下,向四口瓶中投入10g化合物8,2.8g l-脯氨酸,40g 95%乙醇,升温至50℃~60℃,保温30分钟,降温至20℃~25℃,保温12小时,过滤得湿品,控制温度30℃~35℃,真空度0.08~0.10mpa减压干燥36小时,得脯氨酸恒格列净一水合物化合物1干品10g,收率80%。esi-hrms(m/z):c
22
h
25
clfo7[m+h
+
]理论计算值:455.1267,实测值:455.1263。
[0062]
以上公开内容仅是本发明的优选实施方式,从技术层面讲,对于在本发明合成路线构思框架的基础上,对所述的实施步骤中反应条件的若干优化和为获得本发明所涉及的中间体所做出的方法改进也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1