利多卡因中杂质化合物的去除方法及其所得的产物与流程
1.本发明属于药物(或其中间产物)检测和杂质控制领域,具体涉及利多卡因中杂质化合物的去除方法及其所得的产物。
背景技术:
2.药物,包括原料药和制剂,作为预防、治疗和/或诊断疾病的一种特殊商品,其质量的优劣关系着公众的健康和安全。因而,保证其安全有效、质量可控,是贯穿于研发、生产、贮存、临床应用等全部过程中的重中之重。而药物中的各种杂质能否被准确的识别出来并加以控制,直接关系到药物的质量可控性与安全性,是其质量保证的一个关键环节,同时也是药物一致性评价的一项重要指标。
3.杂质,是指由于生产工艺以及原料、助剂等方面的原因而出现在药物中的其他物质。杂质的来源之一,是在药物的生产过程中所引入,可能的原因是:(1)工艺路线及工艺条件的差异;(2)所用的原料不纯;(3)未完全反应;(4)反应的中间产物;(5)发生副反应生成副产物;(6)溶剂、催化剂等助剂的残留;等等。这些引入的杂质,不仅会使药物的纯度降低,而且还会影响到药物的实际疗效,严重者甚至可能引起用药者出现不良反应。过去几十年国内外发生的多个药品安全事件,大多都与药物中的杂质有关。
4.然而,在过去较长的一段时间里该问题并没有引起我们足够的重视,从而导致我国的一些药物品种在疗效、不良反应等方面与进口药品相比仍存在着不小的差距。但随着我国《化学药物杂质研究技术指导原则》(2005年)的颁布,初步确立了药物杂质研究的雏形,这种差距的情况已经逐步在拉近,但总体水平仍落后于欧美发达国家。
5.对于药物中杂质控制这一技术问题,人们的认识基本可概括为三个阶段:药品纯度控制阶段、杂质限度控制阶段和杂质谱控制阶段。过去由于受到技术水平的限制,主要是通过控制api(active pharmaceutical ingredient,原料药)纯度来间接实现药物中杂质的控制,但这种方法的专属性较差,无法实现对各杂质的准确量化,已逐步被杂质限度、杂质谱的控制技术所取代。也就是说,在药物的制备或生产中,杂质限度控制和杂质谱控制代表着这一技术的发展趋势(参见:王维剑.药品杂质控制与评价关键技术研究[d].山东大学博士学位论文,2016:“1.2研究理念”部分)。
[0006]
此外,英国药典(1988年版)还明确指出:“与药品中的己知杂质相比,对未知杂质的发现和控制更加重要。在正常生产条件下,药品中不应当含有量不可控以及性质未知的杂质”。现行的国内外药品生产和质量标准中,基本都是采用有关物质(杂质)、残留溶剂等手段来直接检测杂质的含量(参见:王维剑.药品杂质控制与评价关键技术研究[d].山东大学博士学位论文,2016:“1.2研究理念”部分),进而才可能想方设法的采取技术手段在其制备或生产过程中加以去除和/或控制。
[0007]
具体到利多卡因,化学名称为:n-(2,6-二甲苯基)-2-(二乙氨基)乙酰胺,是一种经过多年临床应用的药物,因具有作用快、安全有效、副作用少等优点,而被广泛用于局部麻醉以及多种原因所引起的室性心律失常等。由于利多卡因本身难溶于水,影响药物的吸
收和生物利用度,因而临床上大多使用的是其盐酸盐和/或水合物,其中比较常见的是盐酸利多卡因一水合物,其结构式如下:
[0008][0009]
盐酸利多卡因一水合物
[0010]
目前,在利多卡因的制备或生产中,杂质控制问题的相关研究仍很粗浅,依旧处于通过控制api纯度来间接实现药物中杂质控制的初始阶段,例如:cn 102070483 a、cn 105294477 a、cn 110590590 a等,这些还无法实现对利多卡因生产工艺杂质的定性和/或定量检测,既不利于在其制备或生产过程中发现新的未知杂质并想办法加以控制,也不利于药物的产品质量保证,进而会影响到公众用药的安全性以及我国医药行业的持续健康发展。
[0011]
有鉴于此,特提出本发明。
技术实现要素:
[0012]
针对现有技术存在的问题和/或不足,本发明的目的在于提供利多卡因中杂质化合物的去除方法。通过该方法,既能够实现利多卡因形成盐酸盐(和/或水合物)改善水溶性和生物利用度,又能够有效地除去利多卡因中少量新发现的杂质化合物,对其进行杂质限度控制,进而保证原料药产品质量可控;而且,工艺条件温和,所得产物的收率和纯度高,杂质含量低,毒副作用小,安全性更高,能够显著提高盐酸利多卡因(和/或其水合物)及其制剂的安全性、有效性和稳定性。
[0013]
本发明提供的利多卡因中式ⅱ所示化合物的去除方法,包括以下步骤:使用或不使用惰性气体保护,用氯化氢气体将利多卡因、盐酸和丙酮的反应液体系的ph值调至2.5~3.8(例如:3.0、3.2、3.4、3.5、3.6等),随后分离纯化,即可;
[0014]
其中,所述的利多卡因中包含式ⅱ所示化合物;
[0015][0016]
式中,r1为羧基,r2为1~3个c1~c3烷基取代的苯基,r3、r4和r5独立地选自h、卤素或nr
11r12
,r
11
和r
12
独立地选自c1~c3烷基。
[0017]
进一步地,
[0018]
在上述利多卡因中式ⅱ所示化合物的去除方法中,某些反应参数可如下所述,未涉及的反应参数如本发明中任一方案所述(以下简称“在上述利多卡因中式ⅱ所示化合物的去除方法中”),所述的式ⅱ所示化合物为式中,r2为2个甲基取代的苯
基;优选地,所述的式ⅱ所示化合物为(杂质b)。
[0019]
进一步地,
[0020]
在上述利多卡因中式ⅱ所示化合物的去除方法中,利多卡因中式ⅱ所示化合物的含量为0.5%~50%(例如:1.0%、1.5%、2%、3%、5%、8%、10%、20%、40%等);优选地,利多卡因中式ⅱ所示化合物的含量为0.5%~5%;更优选地,利多卡因中式ⅱ所示化合物的含量为0.8%~2.5%。
[0021]
进一步地,
[0022]
在上述利多卡因中式ⅱ所示化合物的去除方法中,所述反应液体系的ph值调至3.0~3.8;优选地,所述反应液体系的ph值调至3.4~3.6(即:3.5
±
0.1)。
[0023]
进一步地,
[0024]
在上述利多卡因中式ⅱ所示化合物的去除方法中,每千克利多卡因对应的丙酮用量为1~20l(包括但不限于:2l、3l、5l、8l、10l、15l等);优选地,每千克利多卡因对应的丙酮用量为2.5~5l。
[0025]
在上述利多卡因中式ⅱ所示化合物的去除方法中,所述盐酸的用量可以是常规选择,例如:每千克利多卡因对应的盐酸用量为0.01~0.2kg(包括但不限于:0.02kg、0.05kg、0.1kg、0.12kg、0.13kg、0.15kg等);优选地,每千克利多卡因对应的盐酸用量为0.1~0.14kg。
[0026]
在上述利多卡因中式ⅱ所示化合物的去除方法中,所述盐酸的浓度可以是常规选择,例如:所述盐酸的摩尔浓度为2~12.5mol/l(包括但不限于:3mol/l、5mol/l、8mol/l、10mol/l等);优选地,所述盐酸为浓盐酸。
[0027]
在上述利多卡因中式ⅱ所示化合物的去除方法中,用氯化氢气体调节反应液体系的ph值的过程中控制温度为5~15℃。
[0028]
在上述利多卡因中式ⅱ所示化合物的去除方法中,所述的分离纯化包括以下步骤:将反应液升温至45~60℃(包括但不限于:50~57℃等),然后降温至0~12℃(包括但不限于:2℃、5℃、10℃等),析出固体,分离出目标产物(盐酸利多卡因和/或其水合物),之后以丙酮和水为溶剂重结晶;
[0029]
优选地,重结晶溶剂中丙酮和水的体积比为1:(0.01~0.05),再优选为1:(0.02
±
0.01)(即:1:0.01~0.03);
[0030]
更优选地,所述的重结晶包括以下步骤:使用或不使用惰性气体保护,将分离出的目标产物溶于45~60℃的丙酮和水中,然后降温至30~40℃,过滤,浓缩滤液,补加丙酮,之后降温至0~12℃,析出固体,分离,干燥,即可;其中,每千克目标产物对应的丙酮和水的用量为1~20l(包括但不限于:3l、5l、8l、10l、15l等,该用量是指丙酮和水的总用量),再优选为,每千克目标产物对应的丙酮和水的用量为2~5l。
[0031]
进一步地,
[0032]
在上述利多卡因中式ⅱ所示化合物的去除方法中,所述的利多卡因由包括以下步骤的方法制备而成:
[0033][0034]
式中,x为卤素,优选为氯或溴;
[0035]
使用或不使用惰性气体保护,化合物c与二乙胺发生缩合反应(该反应,既可以在没有碱性物质和溶剂存在的条件下进行,例如:cn 111253273 a等;也可以在没有碱性物质但有溶剂存在的条件下进行,例如:cn 110938012 a等;还可以在碳酸盐和溶剂都存在的条件下进行,例如:cn 102070483 a等),生成利多卡因;优选地,
[0036]
所述的化合物c中包含(杂质a),优选化合物c中的含量为0.5%~1.8%。
[0037]
在上述利多卡因的制备方法中,某些反应参数可如下所述,未涉及的反应参数如本发明任一方案所述(以下简称“在上述利多卡因的制备方法中”),化合物c与二乙胺的摩尔比为1:(1~5)(包括但不限于:1:1.2、1:1.3、1:1.5、1:1.8、1:2、1:2.5、1:3.5等),优选为1:(1.5~3)。
[0038]
在上述利多卡因的制备方法中,所述缩合反应的反应温度为30~60℃,优选为50~57℃。
[0039]
在上述利多卡因的制备方法中,所述缩合反应的反应终点,可通过反应时间和/或原料(化合物c)在反应液中的剩余量(hplc、tlc等方法监测)加以控制和确定,例如:化合物c基本消失(在反应液中的剩余量≤2wt%)作为反应终点。
[0040]
进一步地,
[0041]
在上述利多卡因的制备方法中,化合物c与二乙胺在碳酸盐和缩合溶剂存在的条件下发生缩合反应。
[0042]
在上述利多卡因的制备方法中,所述的碳酸盐为碱金属碳酸盐,优选为碳酸钠或碳酸钾。
[0043]
在上述利多卡因的制备方法中,所述的缩合溶剂(是指用于缩合反应的溶剂)选自丙酮、甲苯或正己烷,优选为丙酮。
[0044]
在上述利多卡因的制备方法中,化合物c与碳酸盐的摩尔比为1:(0.5~5)(包括但不限于:1:0.6、1:0.7、1:1、1:1.2、1:1.5、1:1.8、1:2、1:2.5、1:3.5等),优选为1:(0.75~1.2)。
[0045]
在上述利多卡因的制备方法中,每千克化合物c对应的缩合溶剂用量为1~20l(包括但不限于:2l、3l、5l、8l、10l、15l等),优选为每千克化合物c对应的缩合溶剂用量为1.5~6l。
[0046]
进一步地,
[0047]
在上述利多卡因中式ⅱ所示化合物的去除方法中,所述的化合物c由包括以下步骤的方法制备而成:
[0048][0049]
式中,x为卤素,优选为氯或溴;
[0050]
使用或不使用惰性气体保护,2,6-二甲基苯胺与化合物y在有机溶剂以及碱性物质存在的条件下发生取代反应(2,6-二甲基苯胺上nh2的一个h,被化合物y的卤化乙酰基所取代),生成化合物c。
[0051]
在上述化合物c的制备方法中,某些反应参数可如下所述,未涉及的反应参数如上任一方案所述(以下简称“在上述化合物c的制备方法中”):所述的有机溶剂可以是该类反应的常用溶剂,例如:所述的有机溶剂选自二氯甲烷、丙酮、乙酸、正庚烷、醋酸丁酯、乙酸异丙酯或甲基叔丁基醚等,再优选为二氯甲烷。
[0052]
在上述化合物c的制备方法中,所述的碱性物质为碳酸氢盐、碳酸盐或乙酸盐,再优选为碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾、乙酸钠或乙酸钾。
[0053]
在上述化合物c的制备方法中,2,6-二甲基苯胺与化合物y的摩尔比为1:(1~5)(包括但不限于:1:1.2、1:1.3、1:1.5、1:1.8、1:2、1:2.5、1:3.5等),再优选为1:(1.05~1.5)。
[0054]
在上述化合物c的制备方法中,2,6-二甲基苯胺与碱性物质的摩尔比为1:(0.5~5)(包括但不限于:1:0.6、1:0.7、1:1、1:1.2、1:1.5、1:1.8、1:2、1:2.5、1:3.5等),再优选为1:(0.75~1.2)。
[0055]
在上述化合物c的制备方法中,每千克2,6-二甲基苯胺对应的有机溶剂用量为1~20l(包括但不限于:2l、3l、5l、8l、10l、15l等),再优选为每千克2,6-二甲基苯胺对应的有机溶剂用量为4~10l。
[0056]
在上述化合物c的制备方法中,所述取代反应的反应温度为-8~20℃,再优选为-5~15℃;
[0057]
所述取代反应的反应终点,可通过反应时间和/或原料(2,6-二甲基苯胺)在反应液中的剩余量(tlc等方法监测)加以控制和确定,例如:2,6-二甲基苯胺基本消失(在反应液中的剩余量≤2wt%)作为反应终点。
[0058]
本发明的另一目的在于提供一种安全性高的药物组合物,该药物组合物中的杂质化合物(尤其是杂质b化合物)含量低,不良反应少,有效控制盐酸利多卡因、盐酸利多卡因水合物等原料药或其制剂中杂质化合物的含量,以提高盐酸利多卡因(和/或其水合物)及其制剂的安全性、有效性和稳定性。
[0059]
所述的药物组合物包含盐酸利多卡因水合物和式ⅱ所示化合物,其中,所述盐酸利多卡因水合物的含量不低于99.0%(例如:99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%、99.9%等),所述式ⅱ所示化合物的含量不高于0.25%;
[0060]
[0061]
式中,r1为羧基,r2为1~3个c1~c3烷基取代的苯基,r3、r4和r5独立地选自h、卤素或nr
11r12
,r
11
和r
12
独立地选自c1~c3烷基。
[0062]
所述的药物组合物中,优选地,所述的盐酸利多卡因水合物为盐酸利多卡因一水合物。
[0063]
所述的药物组合物中,优选地,所述的式ⅱ所示化合物为式中,r2为2个甲基取代的苯基;更优选地,所述的式ⅱ所示化合物为(杂质b)。
[0064]
所述的药物组合物中,优选地,所述式ⅱ所示化合物的含量不高于0.05%;更优选地,所述式ⅱ所示化合物的含量不高于0.02%。
[0065]
所述的药物组合物中,优选地,所述的药物组合物是按照前述任意一项所述利多卡因中式ⅱ所示化合物的去除方法所得的产物。
[0066]
本发明中,所述的杂质化合物可以通过以下方法制备得到:
[0067]
例如:杂质a,可以以本发明制得的氯乙酰-2,6-二甲基苯胺作为原料,按照实施例1高效液相色谱检测方法的色谱条件,收集保留时间=22.581min(除非另有说明,一般是指相对保留时间)左右的洗脱液,除去溶剂,即得杂质a化合物;或者,以2,6-二甲基苯胺和二氯乙酰氯(cas号:79-36-7)为原料,按照本领域相同反应或类似反应的常规方法(例如:实施例1中氯乙酰-2,6-二甲基苯胺的制备方法),制备得到杂质a化合物;
[0068]
又如:杂质b,可以以本发明制得的利多卡因作为原料,按照实施例1高效液相色谱检测方法的色谱条件,收集保留时间=9.33min左右的洗脱液,除去溶剂,即得杂质b化合物;或者,按照本领域相同反应或类似反应的常规方法,制备得到杂质b化合物。
[0069]
除非另有说明,本发明中所述的目标产物纯度、杂质含量等这类数据,一般是指hplc检测并按照面积归一化法所计算得到的结果。
[0070]
除非另有说明,本发明中所述的含量通常系指重量百分比含量。
[0071]
本发明的有益效果,具体如下:
[0072]
(1)在利多卡因或其中间体的制备过程中,除了已知杂质以外,还首次检测并鉴定出两种新的杂质化合物:杂质a和杂质b,这为实现技术转型升级为杂质限度控制技术和/或杂质谱控制技术提供了可靠的研究基础;
[0073]
(2)在发现新杂质问题的基础上,提供了解决方案:利用hplc方法实现了对杂质a、杂质b的定性和/或定量检测,专属性强,分离度高,满足准确度和精密度的相关要求,为杂质限度控制技术和/或杂质谱控制技术的顺利实施提供了必要的支持,从而能够更有效的监控利多卡因或其盐、水合物的产品质量,切实保障药物安全;
[0074]
(3)本发明杂质化合物a和/或b的hplc检测方法,各色谱峰之间无明显干扰,分离度高,可以实现对目标产物、杂质化合物的准确检测,便于操作和控制,检测结果准确、可
靠,为监控利多卡因或其中间体、盐、水合物中的新杂质含量提供了一种行之有效的方法;
[0075]
(4)在发现新杂质问题的基础上,提供了解决方案:利用利多卡因与杂质化合物在结构(nhc=o中的h有无被cooh取代)以及性质(nhc=o中的nh与盐酸的成盐能力强,nhc=o中的h被cooh取代之后成盐能力变差)上的差异,既能够实现利多卡因形成其盐酸盐(和/或水合物)改善水溶性和生物利用度,又能够有效除去利多卡因中存在的少量杂质b,对其进行杂质限度控制,进而保证产品质量可控;
[0076]
(5)本发明利多卡因中杂质化合物的去除方法,工艺条件温和,所得产物的收率和纯度高,杂质含量低,毒副作用小,不良反应少,安全性更高,能够显著提高盐酸利多卡因(和/或其水合物)及其制剂的安全性、有效性和稳定性。
附图说明
[0077]
图1是实施例1所得产物氯乙酰-2,6-二甲基苯胺的hplc图。
[0078]
图2是杂质a的1h-nmr图。
[0079]
图3是杂质a的
13
c-nmr图。
[0080]
图4是实施例1所得产物利多卡因的hplc图。
[0081]
图5是杂质b的1h-nmr图。
[0082]
图6是杂质b的
13
c-nmr图。
[0083]
图7是实施例1所得产物盐酸利多卡因一水合物的hplc图。
[0084]
图8是盐酸利多卡因一水合物的1h-nmr图。
[0085]
图9是盐酸利多卡因一水合物的
13
c-nmr图。
[0086]
图10是盐酸利多卡因一水合物的dsc测试结果图。
具体实施方式
[0087]
下面通过实施例的方式对本发明作进一步详细说明,但并不表示因此将本发明的保护范围限制在所述的实施例范围之内。
[0088]
本发明中,未注明具体条件者,按照常规条件或制造商建议的条件进行,所用试剂或仪器未注明生产厂商者,可以通过购买市售商品获得或已知方法制备得到。
[0089]
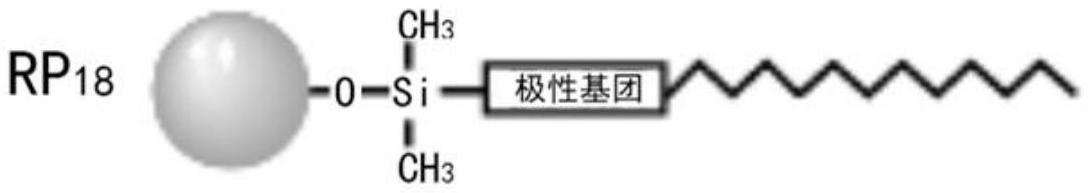
据waters corporation(沃特世公司)官方的相关介绍资料显示,xterra rp 18色谱柱是在烷基和硅胶基质之间嵌入极性基团(极性嵌入型固定相),其示意结构如下:
[0090][0091]
xterra rp 18与xterra ms c18、xterra phenyl、x-bridge、acquity uplc beh等,均属于杂化颗粒柱的一种。
[0092]
参见:
[0093]
1、高效液相色谱柱及消耗品简要指南2008-2009(waters公司).网上公开地址:https://wenku.baidu.com/view/0baf3d4f240c844768eaee62.html,2015/10/17上传。
[0094]
2、液相色谱实战探讨(waters corporation).网上公开地址:https://www.doc88.com/p-9933637355110.html,上传日期:2014/11/05。
[0095]
关于本发明中使用术语的定义,除非另有说明,本文中术语提供的初始定义适用于全文中的该术语;对于本文没有具体定义的术语,应当根据公开内容和/或上下文,给出本领域技术人员能够给予它们的含义。另外,在有些情形下,反应中间产物可以无需分离和/或纯化即可用在随后的步骤中。
[0096]
实施例1
[0097]
1、氯乙酰-2,6-二甲基苯胺的制备
[0098][0099]
包括以下步骤:
[0100]
a、惰性气体(氮气)保护,向反应釜中加入16kg(约132mol)2,6-二甲基苯胺、11.09kg(约132mol)碳酸氢钠和96l二氯甲烷,搅拌均匀,加入16.4kg(约145mol)氯乙酰氯(控制釜内温度-5~15℃),在-5~15℃下搅拌反应1h,gc(气相色谱)或tlc(薄层色谱)监控反应基本完全(2,6-二甲基苯胺的剩余量≤2wt%)后结束;
[0101]
b、向釜内加入16kg纯化水,搅拌均匀,之后减压蒸馏(真空度≤-0.09mpa,温度40~45℃)至基本不出馏分,再向釜内加入176kg纯化水,在15~35℃下搅拌2h后,离心,鼓风干燥(50~60℃),得到氯乙酰-2,6-二甲基苯胺,白色粉末状固体,收率97.88%(以2,6-二甲基苯胺计)。
[0102]
对上述产物氯乙酰-2,6-二甲基苯胺进行高效液相色谱(hplc)检测,色谱条件如下:
[0103]
色谱柱:waters xterra rp 18,4.6mm
×
250mm,5μm;
[0104]
流动相:4.85g/l磷酸二氢钾水溶液(用氢氧化钠调节ph至8.0)-甲醇-乙腈(60:20:20);
[0105]
流速:1.0ml/min;
[0106]
uv检测器(检测波长230nm);
[0107]
柱温:30℃;
[0108]
溶剂:水-乙腈(1:1);
[0109]
进样体积为20μl;
[0110]
hplc检测结果见图1,氯乙酰-2,6-二甲基苯胺(保留时间=10.188min)纯度为98.98%,杂质a(保留时间=22.581min)的含量为1.00%;该检测方法符合中国药典(2015年版)中准确度和精密度的相关要求,专属性强,分离度高。
[0111]
杂质a的1h-nmr(cdcl3,见图2))、
13
c-nmr(cdcl3,见图3)和ir检测结果,分别见表1~3。
[0112]
表1、杂质a的1h-nmr(cdcl3)检测数据
[0113][0114]
表2、杂质a的
13
c-nmr(cdcl3)检测数据
[0115][0116]
表3、杂质a的ir检测数据
[0117]
吸收波数(cm-1
)归属2924、2857甲基c-h伸缩振动1470甲基c-h面内摇摆1379甲基c-h剪式振动1541、1604苯环c=c伸缩振动3038、3008苯环c-h伸缩振动707c-cl伸缩振动1675酰胺c=o伸缩振动3442n-h伸缩振动
[0118]
杂质a的ms检测:m+h
+
=231.94。
[0119]
杂质a的uv检测:在乙腈-水(1∶1)溶液中的最大吸收波长为194.80nm。最终确定,杂质a的结构式如下:
[0120][0121]
2、利多卡因的制备
[0122][0123]
包括以下步骤:
[0124]
i、惰性气体(氮气)保护,向反应釜中加入24kg(约121.5mol)氯乙酰-2,6-二甲基
苯胺(前一工序所得)、17.76kg(约242.8mol)二乙胺、13.43kg(约97.2mol)碳酸钾和120l丙酮,在50~57℃下搅拌反应10h,hplc(高效液相色谱)或tlc监控反应基本完全(氯乙酰-2,6-二甲基苯胺的剩余量≤2wt%)后结束;
[0125]
ii、将反应釜中的反应液降温至20~30℃,抽滤,滤饼用丙酮淋洗两次(5kg
×
2),得到滤液,减压蒸馏(真空度≤-0.09mpa,温度40~45℃)至基本不出馏分,加入96kg纯化水,在15~35℃下搅拌5h后,离心,鼓风干燥(40~50℃),得到利多卡因,白色粉末状固体,收率95.05%(以氯乙酰-2,6-二甲基苯胺计)。
[0126]
对上述产物利多卡因进行高效液相色谱(hplc)检测,色谱条件如下:
[0127]
色谱柱:waters xterra rp 18,3.9mm
×
150mm,5μm;
[0128]
流动相:4.85g/l磷酸二氢钾水溶液(用氢氧化钠调节ph至8.0)-乙腈(70:30);
[0129]
流速:1.0ml/min;
[0130]
uv检测器(检测波长230nm);
[0131]
柱温:30℃;
[0132]
溶剂:流动相;
[0133]
进样体积为20μl;
[0134]
hplc检测结果见图4,利多卡因(保留时间=14.913min)纯度为97.95%,杂质b(保留时间=9.330min)的含量为1.63%,氯乙酰-2,6-二甲基苯胺(保留时间=5.520min)的含量为0.23%;该检测方法符合中国药典(2015年版)中准确度和精密度的相关要求,专属性强,分离度高。
[0135]
杂质b的1h-nmr(cdcl3,见图5)、
13
c-nmr(cdcl3,见图6)和ir检测结果,分别见表4~6。
[0136]
表4、杂质b的1h-nmr(cdcl3)检测数据
[0137][0138]
表5、杂质b的
13
c-nmr(cdcl3)检测数据
[0139][0140]
表6、杂质b的ir检测数据
[0141]
吸收波数(cm-1
)归属2966、2930甲基c-h伸缩振动1461甲基c-h面内摇摆1380甲基c-h剪式振动1537、1603苯环c=c伸缩振动3048苯环c-h伸缩振动1675酰胺c=o伸缩振动
[0142]
杂质b的ms检测:m+na
+
=301.00。
[0143]
杂质b的uv检测:在乙腈溶液中的最大吸收波长为194.40nm。最终确定,杂质b的结构式如下:
[0144][0145]
3、盐酸利多卡因一水合物的制备与纯化
[0146][0147]
包括以下步骤:
[0148]
①
、惰性气体(氮气)保护,向反应釜中加入26kg利多卡因(前一工序所得)、3.17kg浓盐酸(浓度36%~38%)、78l丙酮,搅拌溶解后,通入氯化氢气体(约加入2.8kg的hcl)至釜内液体的ph值检测为3.5
±
0.1结束,此过程中釜内液体温度控制在5~15℃;
[0149]
②
、将反应釜中的反应液升温至50~57℃搅拌2h,之后降温至0~10℃,继续搅拌8h,离心,真空干燥(真空度≤-0.09mpa,温度40~45℃),得到粗品;
[0150]
进一步纯化:
[0151]
惰性气体(氮气)保护,将27kg粗品加入到81l丙酮和1.68kg纯化水中,升温至50~57℃搅拌2h,之后降温至30~40℃,抽滤,滤饼用丙酮淋洗两次(5kg
×
2),得到滤液,减压蒸馏(真空度≤-0.09mpa,温度25~35℃)至基本不出馏分,加入54l丙酮,搅拌均匀,之后降温至0~10℃继续搅拌5h,离心,真空干燥(真空度≤-0.09mpa,温度30~50℃),得到盐酸利多卡因一水合物,白色粉末状固体,收率为81.5%,hplc检测结果见图7,产品纯度99.98%,杂质b的含量≤0.02%,熔点75.5~78.5℃,含水量检测值6.3%(理论值6.23%)。
[0152]
对制得的盐酸利多卡因一水合物进行如下检测:
[0153]1h-nmr(d2o)检测结果,如图8所示;
13
c-nmr(d2o)检测结果,如图9所示;dsc(differential scanning calorimetry,差示扫描量热法)测试结果,如图10所示。
[0154]
x-射线衍射检测结果(仪器型号:xd-6型x-射线衍射仪,起始角:3
°
,终止角:60
°
,扫描速度8
°
/分,采样步宽:0.01,高压设定:36kv,电流:20ma,功率:1.5kw;peak:41-pts/
parabolic filter,threshold=5.0,cutoff=10.0%,bg=5/1.0,peak-top=summit),见表7。
[0155]
表7、盐酸利多卡因一水合物的x-射线衍射检测结果
[0156][0157][0158]
同时,结合元素分析的数据结果:c 58.58%(理论值58.22%)、h 8.704%(理论值8.73%)、n 9.45%(理论值9.70%),最终确定得到的是盐酸利多卡因一水合物。
[0159]
实施例2和3
[0160]
与实施例1相同的内容不再重复,不同的是,制备氯乙酰-2,6-二甲基苯胺的步骤a中,将氯乙酰氯的用量分别变更为132mol、158.4mol,即:1当量、1.2当量,最终得到氯乙酰-2,6-二甲基苯胺,白色粉末状固体,收率均≥90%(以2,6-二甲基苯胺计),hplc纯度均≥98%,杂质a的含量在0.6%~1.5%范围内。
[0161]
实施例4~6
[0162]
与实施例1相同的内容不再重复,不同的是,制备氯乙酰-2,6-二甲基苯胺的步骤a中,将碳酸氢钠的用量分别变更为106mol、158.4mol、198mol,即:0.8当量、1.2当量、1.5当量,最终得到氯乙酰-2,6-二甲基苯胺,白色粉末状固体,收率均≥95%(以2,6-二甲基苯胺计),hplc纯度均≥98%,杂质a的含量在0.6%~1.5%范围内。
[0163]
实施例7和8
[0164]
与实施例1相同的内容不再重复,不同的是,制备利多卡因的步骤i中,将二乙胺的用量分别变更为121.5mol、364.5mol,即:1当量、3当量,最终得到利多卡因,白色粉末状固体,收率分别为77.4%、96.5%(以氯乙酰-2,6-二甲基苯胺计),hplc纯度均≥98%,杂质b
的含量在1.0%~2.5%范围内。
[0165]
实施例9和10
[0166]
与实施例1相同的内容不再重复,不同的是,制备利多卡因的步骤i中,将碳酸钾的用量分别变更为48.6mol、121.5mol,即:0.4当量、1当量,最终得到利多卡因,白色粉末状固体,收率≥80%(以氯乙酰-2,6-二甲基苯胺计),hplc纯度均≥98%,杂质b的含量在1.0%~2.5%范围内。
[0167]
实施例11
[0168]
与实施例1相同的内容不再重复,不同的是,制备利多卡因的步骤i中,将反应温度调整为30~35℃,最终得到利多卡因,白色粉末状固体,收率≥80%(以氯乙酰-2,6-二甲基苯胺计),hplc纯度均≥90%,杂质b的含量在1.0%~2.5%范围内,氯乙酰-2,6-二甲基苯胺的含量在7.0%~9.5%范围内。
[0169]
实施例12~14
[0170]
与实施例1相同的内容不再重复,不同的是,制备盐酸利多卡因一水合物的步骤
①
中,将通入氯化氢气体的用量分别调整为釜内液体的ph值为1.0
±
0.1、2.0
±
0.1、4.0
±
0.1,最终得到制备盐酸利多卡因一水合物,白色粉末状固体,其收率、hplc纯度、杂质b的含量见表8。
[0171]
表8、通入氯化氢气体调整ph值的影响
[0172] 实施例12实施例13实施例1实施例14调整ph值1.0
±
0.12.0
±
0.13.5
±
0.14.0
±
0.1收率50.2%59.1%81.5%68.5%hplc纯度≥98%≥98%99.98%≥98%杂质b的含量未检测未检测≤0.02%0.23%
[0173]
虽然以上描述了本发明的具体实施方式,但是本领域的技术人员应当理解,这仅是举例说明,本发明的保护范围是由所附权利要求书限定的。本领域的技术人员在不背离本发明的原理和实质的前提下,可以对这些实施方式做出多种变更或修改,但这些变更和修改均落入本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1