一种新型吡嗪结构FXR激动剂、制备方法及应用与流程
一种新型吡嗪结构fxr激动剂、制备方法及应用
技术领域
1.本发明涉及一种药物化合物的制备方法,特别涉及一种新型吡嗪结构fxr激动剂、制备方法及应用。
背景技术:
2.自1999年发现胆汁酸能激活fxr产生多种生理功能以来,具有选择性和高活性的fxr激动剂相继被发现,这些fxr激动剂按结构可分为甾体类和非甾体类。甾体类主要为鹅脱氧胆碱(chenodeoxycholic acid,cdca,胆汁酸其中一种)及其衍生物和merck公司开发的fxr激动剂mfa-1;非甾体类包括异噁唑类化合物gw4064及其类似物,fexaramine类化合物,氮杂并吲哚类化合物xl335及其衍生物,苯咪唑基酰胺类化合物,吡唑啶二酮类化合物等。
3.非酒精性脂肪性肝病(non-alcoholic fatty liver disease nafld)是一组无过量饮酒史,肝组织学改变与酒精性肝病相类似,以肝实质细胞脂肪变性和脂肪贮积为特征的临床病理综合征,包括单纯性脂肪肝以及由其演变的脂肪性肝炎和肝硬化。现今nafld已成为仅次于乙肝的影响人类健康的第二类慢性肝脏疾病。nash是一种肝内脂肪积聚而导致的慢性进展性肝病,可导致肝硬化、肝衰竭及肝细胞癌,确切的说,nash只是非酒精性脂肪性肝病(nafld)病程发展的一个阶段。
4.临床在研药物中,奥贝胆酸是一种fxr激动剂。在2016年5月被fda批准,用于治疗原发性胆汁酸肝硬化(pbc),也是第一个进入iii期临床的nash药物。奥贝胆酸针对伴有2~3级肝纤维化的nash患者的关键iii期regenerate研究的期中分析取得积极结果。
5.其他的fxr激动剂如px-104已经进入ii期临床,主要适应症也是非酒精性脂肪肝病(nafld)。
[0006][0007]
2000年maloney等报道了第一个具有高活性和高选择性的异噁唑类fxr激动剂gw4064,其胞外活性为ec
50
=15nmol
·
l-1
,在细胞水平的ec
50
值为90nmol
·
l-1
,可以完全激动fxr靶点蛋白;但在药代动力学方面,在t1/2=3.5小时的口服利用度只有10%,不具备成药的条件,所以作为研究fxr功能和相关疾病的工具化合物。随后gsk、novartis、roche、lilly、phenex等公司分别对gw4064的结构进行了改造,以求得到活性更高,口服利用度更好,更具成药性质的新化合物。到目前为止,数目众多的异噁唑类化合物被成功合成,但在活性、水溶性及口服生物利用度等成药性方面均有不足,
[0008][0009]
中国专利cn103702719a公开了新型fxr(nr1h4)结合及活性调节化合物,其中具体公开了如下化合物:
[0010][0011]
以及这些化合物在治疗非酒精性脂肪肝病(nafld)或非酒精性脂肪性肝炎(nash)等疾病中的应用。这些化合物是在wo2011/020615公开的化合物的基础上进行的改进,通过替换1,2-环亚丙基的1,3-环亚丁基或1,3-亚氮杂环丁烷基上引入极性羟基实现的。结果表明,所得化合物保持了在fxr受体上的活性,而且显现出改进的物化性能,例如较高的水溶性和/或膜渗透性。而更好的水溶性和膜渗透性导致更高的口服生物利用度。但是,上述化合物在法尼醇核受体fxr靶点的激活活性相对较低。
技术实现要素:
[0012]
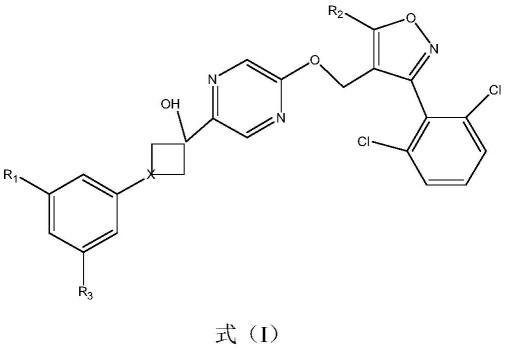
为了解决上述技术问题,本发明提供一种具有式(i)结构的化合物,或其药学上可接受的盐,水合物、溶剂化物、药学上可接受的盐或其拆分的单一异构体:
[0013][0013][0014]
其中,r1选自:卤素、-cooh、
[0015]
r2选自:c
1-c6的烃基、环烃基、芳香基、取代烃基或取代芳香基;
[0016]
r3选自:-h或c1~c3的烃基、环烃基、取代烃基;
[0017]
x为c或n。
[0018]
优选的,其中,
[0019]
r1选自-br;
[0020]
r2选自c
1-c3的烃基、环烃基;
[0021]
r3选自-ch3;
[0022]
x为c或n。
[0023]
更优选的,其中,
[0024]
r2选自
[0025]
最优选的选自以下化合物为:
[0026]
[0027][0028]
本发明进一步提供以所述的化合物作为活性成分的药物组合物。
[0029]
所述的药物组合物,根据需要,还可含有药学上可接受的载体。
[0030]
本发明进一步提供所述的化合物的制备方法,包括如下步骤:
[0031]
本发明所述的化合物在制备治疗非酒精性脂肪肝的药物中的用途。
[0032]
其中所述非酒精性脂肪肝,为非酒精性脂肪肝炎。
[0033]
本发明所述药物组合物在制备治疗非酒精性脂肪肝的药物中的用途,优选非酒精性脂肪肝炎。
[0034]
本发明所述化合物包括其所有异构体形式和异构体混合物的形式。也可以以溶剂化物的形式存在。
[0035]
本发明的药物组合物,优选的是单位剂量的药物制剂形式,在制成药物制剂时可以制成任何可药用的剂型,这些剂型选自:片剂、糖衣片剂、薄膜衣片剂、肠溶衣片剂、胶囊剂、硬胶囊剂、软胶囊剂、口服液、口含剂、颗粒剂、混悬剂、溶液剂、注射剂、栓剂、软膏剂、硬膏剂、霜剂、喷雾剂、贴剂。优选的是口服制剂形式,最佳优选的是片剂,胶囊剂。
[0036]
可以采用制剂学常规技术制备该药物制剂,所述药学上可接受的的载体包括但不限于:甘露醇、山梨醇、山梨酸或钾盐、焦亚硫酸钠、亚硫酸氢钠、硫代硫酸钠、盐酸半胱氨酸、巯基乙酸、蛋氨酸、维生素a、维生素c、维生素e、维生素d、氮酮、edta二钠、edta钙钠,一价碱金属的碳酸盐、醋酸盐、磷酸盐或其水溶液、盐酸、醋酸、硫酸、磷酸、氨基酸、富马酸、氯化钠、氯化钾、乳酸钠、木糖醇、麦芽糖、葡萄糖、果糖、右旋糖苷、甘氨酸、淀粉、蔗糖、乳糖、硅衍生物、纤维素及其衍生物、藻酸盐、明胶、聚乙烯吡咯烷酮、甘油、丙二醇、乙醇、吐温60-80、司盘-80、蜂蜡、羊毛脂、液体石蜡、十六醇、没食子酸酯类、琼脂、三乙醇胺、碱性氨基酸、尿素、尿囊素、碳酸钙、碳酸氢钙、聚乙二醇、环糊精(如β-环糊精)、磷脂类材料、高岭土、滑石粉、硬脂酸钙、硬脂酸镁等。
[0037]
本发明的药物组合物,在制成药剂时,单位剂量的药剂可含有本发明的药物活性物质0.1-1000mg,其余为药学上可接受的载体。药学上可接受的载体以重量计可以是制剂总重量的0.1-99.9%。
[0038]
本发明的药物组合物在使用时根据病人的情况确定用法用量。
[0039]
本发明优选的制备方法包括以下步骤:
[0040]
1)异噁唑中间体合成方法为,苯甲醛1a与盐酸羟胺在碱性条件下缩合得到苯甲醛
肟1b,肟1b在ncs/dmf条件下生成氯代苯甲醛肟1c;接着与酮酸酯通过1,3-偶极环加成反应得到三取代异噁唑环1d-1f;氮气保护下加入dibal-h,还原酯键为羟基,得到中间体1-a-1-c;
[0041]
2)异噁唑中间体与二溴吡嗪在碱性条件下发生亲核取代反应,得到吡嗪中间体2-a-2-c;控制温度保持在零下78℃,用正丁基锂进行锂溴交换与四元环2a-d进行碳-碳键的连接,生成末端酯基产物3-a-3-g,最后碱性条件下发生酯水解反应得到目标化合物tm。
[0042]
本发明的有益效果:本发明的化合物尤其是tm-01和tm-09对肝脏甘油三酯和胆固醇水平有明显的降低作用,提高了小鼠肝重和肝脏体重比,降低了病理评分和肝脏中的胶原沉积,对非酒精性脂肪肝有改善效果。
[0043]
本发明的化合物对法尼醇核受体fxr靶点的具有较强激动活性,并表现出了在细胞膜间的高通透性,且在高脂饲喂小鼠模型中,对脂肪富集的肝脏重量呈现剂量依赖影响,预测在肥胖、糖尿病和非酒精性脂肪肝(nash)等代谢类疾病的治疗中表现出较好的用途,以下通过实验数据说明本发明的有益效果。
[0044]
试验例一、本发明化合物对fxr受体的激动活性研究
[0045]
实验步骤:fxr(是gst标记的重组人fxr蛋白,厂家invitroren,货号pv4835)和src-1(类固醇受体共激活因子-1)在冰上进行融化,用缓冲液配制abc三种溶液,a液,0.4n m fxr和30nm src-1;b液,10ug/ml acceptor beads(受体微珠);c液,10ug/ml donor beads(供体微珠)。将a液加入到板中,每孔15ul。室温孵育1小时。将b液加入到板中,每孔7.5ul。室温孵育1小时。将c液加入板中,每孔7.5ul。室温孵育1小时。酶标仪envision读数。数据用prism 5.0进行曲线拟合,计算出ec
50
。结果见表1。
[0046]
表1
[0047]
[0048]
[0049][0050]
根据表1中的样品活性结果,去除活性较差的去除tm-05、tm-07、tm-10,选择其余7个样品进行了细胞内fxr-tr-fret实验。
[0051]
试验例二、细胞内fxr-tr-fret实验
[0052]
1、细胞培养:a、用适当的密度在10ml完整的胰蛋白酶中胰蛋白酶消化皿和种子细胞中在37℃。b、5%的co2和潮湿条件下培养细胞24小时。
[0053]
2、细胞接种和转染:fugene hd转染试剂用作转染试剂。
[0054]
a根据以下流程准备转染混合物:
[0055][0056]
b用力敲打试管以混合内容物。将混合物在室温下孵育15分钟。
[0057]
c用胰蛋白酶消化盘并确定细胞密度。
[0058]
d将细胞浆以500,000个细胞/ml的密度稀释至所需的体积(对于96孔板,为100ul/孔)。
[0059]
e将所需体积的先前制备的转染混合物添加到两个细胞浆液中,然后将100ul/孔的细胞浆液分配到测定板上。
[0060]
f在加湿条件下于37℃,5%co2下将测定板孵育24小时。
[0061]
3、化合物配制:
[0062]
a、准备fxr工作浓度为10mm的化合物库存,然后在100%dmso中稀释3倍。
[0063]
b、将10ul化合物添加到90ul完全培养基中。
[0064]
c、向每个孔中加入5ul化合物溶液。
[0065]
d、将板在37℃,5%co2和湿润条件下孵育18小时。
[0066]
4、双重荧光素酶测定:萤火虫和海肾荧光素酶信号通过promega的dual luciferase reporter assay system进行分析。envision用作光度计。
[0067]
5、结果计算:通过将萤火虫信号除以海肾信号来归一化数据值。“f/r”表示“萤火虫/renilla”。这种标准化消除了每个孔中不同细胞数量和转染效率的差异。计算%activation(活性)值。通过以下公式计算%activation值。
[0068][0069]
x是每个浓度点的“f/r”值。最小值是无化合物对照的平均“f/r”值。最大值是参考化合物对照的平均值“f/r”值。
[0070]
6、活性结果见表2,
[0071]
表2:活性结果
[0072]
样品编号ec
50
(μm)tm-011.28tm-023.14tm-03》5000tm-04》5000tm-061.79tm-081.51tm-090.26gw40640.30m31.49
[0073]
对受试化合物对于fxr-tr-fret的活性进行了研究,此类含吡嗪结构的化合物,相比对照品gw4064和m3,多数表现出了细胞水平上较好的激动活性。
[0074]
根据fxr-tr-fret实验结果,去除tm-03和tm-04,对剩余部分化合物进行caco-2单层细胞膜转运实验。
[0075]
试验例三、caco-2单层细胞膜转运实验
[0076]
利用人源性结肠腺癌细胞系caco-2单层细胞模型研究目标化合物由绒毛面侧(apical,ap)到基底面侧(basolateral,bl)以及从bl侧到ap侧的双向转运情况,应用高效液相色谱法定量分析,计算转运参数和表观渗透系数(apparent permeabilitycoefficient,papp)以及外排率(efflux ratio),以m3为阳性对照,以p-gp的作用底物地高辛为参照物,选取这5个细胞内活性较高的样品进行试验,来预测这类吡嗪结构异噁唑类衍生物的体内口服生物利用度以及与p-gp的亲和作用情况。
[0077]
结果见表3、4、5
[0078]
表3:在caco-2细胞模型中的a-to-b的表观渗透系数
[0079][0080][0081]
表4:在caco-2细胞模型中的透膜质量回收率
[0082][0083]
表5在caco-2细胞模型中的外排率
[0084][0085]
注:a外排率=papp b-a/papp a-b
[0086]
实验结果如表3所示,本发明的这一系列吡嗪结构异噁唑类化合物a-to-b的papp值都高于p-gp的底物地高辛(papp a-to-b《0.04),优于m3(papp a-to-b《0.15),特别是tm-01、tm-02,tm-06和tm-09这四个化合物的papp,a-to-b值》2.5
×
10-6cm/s,属于高通透性底物。这些数据表明这类吡嗪结构异噁唑类化合物具有良好的通膜能力,预测在体内的吸收优于m3。
[0087]
表4中显示的是这些含吡嗪结构化合物的透膜后的回收率情况。对这5个化合物对其双向转运情况进行评价,结果如表5所示,从外排率上可以看到相对于m3来说,这类衍生物都很大程度上减弱了外排情况,其外排率均远远小于对照品地高辛(外排率》262.93),预测在体内的口服吸收也会相应得到提高。
[0088]
根据试验例三结果,去除效果较差的tm-08,保留其他化合物进行动物实验,关于对照组:m3因为透膜效果较差,被排除。gw4064因为报道成药性较差,生物口服利用度较低,目前作为前期研究的一个工具分子,被排除。m2也是作为前期一个对照化合物,在首次蛋白水平的筛选中活性低于本专利中化合物,被排除。奥贝胆酸是目前非酒精性脂肪领域处在临床研究的药物,接近获批,选取作为动物试验的对照品。
[0089]
试验例四:高脂饲料饲喂(mcd)模拟非酒精性脂肪肝的药效试验
[0090]
奥贝胆酸是目前非酒精性脂肪领域处在临床研究的药物,接近获批,选取作为动物试验的对照品。
[0091]
1、实验动物
[0092]
动物品系:c57/bl6,动物等级:spf级,性别:雄性,动物年龄:8周龄,动物接受日期:2018年09月28日,动物来源:上海灵畅生物科技有限公司,动物合格证号:scxk(沪)2013-00182013001836799,小鼠饲养环境:温度20-26℃,湿度40-70%,12小时昼夜循环。
[0093]
2、试验设计与方法
[0094]
2.1药物配置
[0095]
2.1.1溶媒配制:
[0096]
5%solutol hs15/生理盐水:solutol hs15(聚乙二醇15羟硬脂酸酯增溶剂)在37℃水浴锅中溶化后,取5ml溶于100ml生理盐水中,充分搅拌后备用。
[0097]
2.1.2给药溶液配制:
[0098]
奥贝胆酸/5%solutol hs15/生理盐水:精确称取12mg奥贝胆酸,加入0.15ml solutol hs15使之充分溶解,加入2.85ml生理盐水,剧烈涡旋,超声助溶使其充分溶解。
[0099]
tm-01组/5%solutol hs15/生理盐水:取2.5ml 6mg/ml的tm-01悬浮液,加入2.5ml 5%solutol hs15/生理盐水,充分混匀。
[0100]
tm-02组/5%solutol hs15/生理盐水:取2.5ml 6mg/ml的tm-02悬浮液,加入2.5ml 5%solutol hs15/生理盐水,充分混匀。
[0101]
tm-06组/5%solutol hs15/生理盐水:取2.5ml 6mg/ml的tm-06悬浮液,加入2.5ml 5%solutol hs15/生理盐水,充分混匀。
[0102]
tm-09组/5%solutol hs15/生理盐水:取2.5ml 6mg/ml的tm-09悬浮液,加入2.5ml 5%solutol hs15/生理盐水,充分混匀。
[0103]
所有药物给药前新鲜配制。
[0104]
2.2给药途径及给药容积
[0105]
溶媒与测试物灌胃给药,给药体积为10ml/kg;
[0106]
2.3实验过程、分组和具体给药方式
[0107]
70只8周龄的c57/bl6小鼠到达动物房后,进行适应喂养,待平均体重到达23g后,根据体重将小鼠随机分成7组,并更换成模型饲料。第一组给予对照饲料mcs,其余组给予mcd粮食,同时各组小鼠按以下方式接受化合物处理:
[0108]
组1对照组:mcs,溶媒,每天一次灌胃给药;
[0109]
组2模型组:mcd,溶媒,每天一次灌胃给药;
[0110]
组3奥贝胆酸组:mcd,40mpk奥贝胆酸,每天一次灌胃给药;
[0111]
组4.tm-01组:mcd,30mpk tm-01,每天一次灌胃给药;
[0112]
组5.tm-02组:mcd,30mpk tm-02,每天一次灌胃给药;
[0113]
组6.tm-06组:mcd,30mpk tm-06,每天一次灌胃给药;
[0114]
组7.tm-09组:mcd,30mpk tm-09,每天一次灌胃给药;
[0115]
2.4实验方法
[0116]
在实验过程中,每周测定体重和摄食量。给药21天后,用微量毛细管进行尾尖取血进行ast,alt测定。给药28天后,终止小鼠,心脏取血,收集肝组织,称重,一部分用液氮速冻用于后续分析,另一部分固定进行病理学分析。
[0117]
2.4.1血液指标测定
[0118]
小鼠血液在5000rpm离心10分钟,收集上清,用于tg(甘油三酯),tc(总胆固醇),hdl(高密度脂蛋白),ldl(低密度脂蛋白),ast(谷草转氨酶)和alt(谷丙转氨酶)的测定。
[0119]
给药三周和四周后的ast和alt按试剂盒说明书进行测定;给药4周后的血液脂质指标送艾迪康医学检验所有限公司检测。
[0120]
2.4.2血液细胞因子的测定
[0121]
小鼠血液在5000rpm离心10分钟,收集上清,用于细胞因子(mkc和mcp1)的检测。检测前血清储存于-80℃。
[0122]
mkc和mcp1的测定按试剂盒说明书进行。
[0123]
2.4.3肝脏tc和tg的测定
[0124]
肝脏从-80℃取出后,pbs(磷酸盐缓冲生理盐水)中匀浆,氯仿甲醇有机相抽提后,
用试剂盒进行tc和tg的测定,用蛋白量进行归一化。
[0125]
2.4.4组织收集
[0126]
给药4周后,麻醉小鼠,心脏取血后,分离肝脏称重,取右侧叶用液氮速冻,保存于-80℃用于肝脏脂质分析。分离左侧叶,于10%福尔马林中固定,用于后续的he和sirius red染色。
[0127]
2.5结果处理和数据分析
[0128]
试验结果以均数
±
标准误(mean
±
sem)表示,用t-test进行显著性分析。与模型组比较,*表示p《0.05为有显著性差异,**表示p《0.01为有强显著性差异,***p《0.001为有极显著性差异。
[0129]
3试验结果
[0130]
3.1化合物对小鼠体重和摄食量的影响
[0131]
小鼠经mcd喂养后,体重即如预期显著性下降。并且随着时间的延长,体重持续下降。经化合物处理的各实验组较模型组体重也略有下降(表6)。
[0132]
表6:化合物对小鼠体重的影响
[0133][0134][0135]
表7:化合物对小鼠摄食量的影响
[0136][0137]
3.2化合物对血液指标的影响
[0138]
mcd和化合物处理3周后,取尾尖血进行ast和alt测定。结果表明,模型组的alt较对照组升高了约3倍,ast升高了2倍以上,奥贝胆酸组导致了比模型组更高的ast和alt水平。tm-01组、tm-06组和tm-09组的alt和ast较模型组有显著下降,tm-02组与奥贝胆酸组表现一致。(表8)
[0139]
mcd和化合物处理4周后,模型组中动物血脂水平略高于对照组。奥贝胆酸处理降
低了血液中的胆固醇含量。tm-01组、tm-02组、tm-06组和tm-09组对血脂有不同的降低作用(表9)。
[0140]
表8:化合物处理3周和4周后ast和alt变化
[0141][0142]
表9:化合物处理4周后血脂水平变化
[0143]
组别tg(mmol/l)tc(mmol/l)ldl(mmol/l)hdl(mmol/l)组1-control0.52
±
0.10*0.77
±
0.09***2.05
±
0.06***0.34
±
0.03**组2-model+vehicle0.60
±
0.031.12
±
0.082.90
±
0.040.60
±
0.01组3-obeticholic acid0.56
±
0.020.86
±
0.04**2.80
±
0.030.55
±
0.01**组4-tm-01:30mpk0.53
±
0.100.95
±
0.44*2.61
±
0.26*0.59
±
0.11组5-tm-02:30mpk0.57
±
0.061.25
±
0.142.93
±
0.120.61
±
0.02*组6-tm-06:30mpk0.59
±
0.020.97
±
0.163.01
±
0.09*0.42
±
0.06***组7-tm-09:30mpk0.54
±
0.04*0.86
±
0.09*2.52
±
0.07**0.56
±
0.02*
[0144]
3.3化合物对肝脂含量的影响
[0145]
mcd和化合物处理4周后,收集肝脏测定脂质,肝脂含量用蛋白进行均一化。在模型组中,肝脏甘油三酯和胆固醇显著高于对照组。奥贝胆酸处理后,这两个指标均略有所下降。tm-01组、tm-02组、tm-06组和tm-09组对肝脏甘油三酯(liver tc)和总胆固醇的含量(liver tg)有不同的降低效果,其中tm-01组和tm-09组表现出显著降低水平。(表10)。
[0146]
表10:化合物处理后肝脏脂含量变化
[0147]
组别liver tg(mol/g protein)liver tc(mol/g protein)组1-control112.55
±
8.59***21.47
±
1.29***组2-model+vehicle339.42
±
40.2444.94
±
5.50组3-obeticholic acid298.46
±
36.2535.7
±
3.38组4-tm-01:30mpk246.69
±
54.09*34.85
±
8.20组5-tm-02:30mpk307.38
±
26.3041.67
±
5.73组6-tm-06:30mpk303.5
±
46.5238.23
±
5.04组7-tm-09:30mpk231.73
±
18.4*37.67
±
3.65*
[0148]
3.4化合物对小鼠肝重的影响
[0149]
mcd和化合物处理4周后,收集肝脏,称重。模型组小鼠的肝重和肝脏体重比均显著低于对照组。分析结果时意外的发现此类化合物对肝重有明显的影响,其中tm-01组、tm-02组、tm-06组和tm-09组均显著提高了小鼠肝重(liver weight)和肝脏体重比(即liver/
body weight)。(表11)。
[0150]
表11:化合物处理对肝重的影响
[0151][0152]
3.5化合物肝脏病理的影响
[0153]
小鼠肝脏固定后,进行he和sirius red染色。染色结束后,全片扫描。随机选取6个20x视野。he染色的6个20x视野综合进行病理评分(即he score)。病理评分标准如表12。sirius red染色的6个20x视野图像用image j计算sirius red的阳性染色面积与总面积比(即sirius red staining area)。
[0154]
表12:病理评分标准
[0155][0156][0157]
经mcd喂养4周后,模型组肝脏中有脂肪细胞堆积,间或可见炎性细胞浸润。奥贝胆酸处理后,脂肪细胞堆积和炎性细胞浸润更为显著。病理评分显示,mcd喂养后,模型组病理评分在2左右,奥贝胆酸处理后评分有所升高,而化合物处理后,病理状况有一定的减轻(表13)。
[0158]
表13:病理评分结果
[0159][0160]
3.4结论:mcd喂养小鼠4周后,诱导了血液中ast,alt和脂质的上升,促使脂肪在肝脏中的堆积和炎性灶的生成,建模具有非酒精性脂肪肝的各项指标特征。
[0161]
本发明提供的化合物尤其是tm-01和tm-09对肝脏甘油三酯和胆固醇水平有明显的降低作用,提高了小鼠肝重和肝脏体重比,并在一定程度上降低了病理评分和肝脏中的胶原沉积,初步判定在动物体内对非酒精性脂肪肝有改善效果。
[0162]
说明书附图
[0163]
图1为本发明化合物tm-1的1h-nmr谱图;
[0164]
图2为本发明化合物tm-1的质谱图;
[0165]
图3为本发明化合物tm-2的1h-nmr谱图;
[0166]
图4为本发明化合物tm-2的质谱图;
[0167]
图5为本发明化合物tm-3的1h-nmr谱图;
[0168]
图6为本发明化合物tm-3的
13
c-nmr谱图;
[0169]
图7为本发明化合物tm-3的质谱图;
[0170]
图8为本发明化合物tm-4的1h-nmr谱图;
[0171]
图9为本发明化合物tm-4的质谱图;
[0172]
图10为本发明化合物tm-5的1h-nmr谱图;
[0173]
图11为本发明化合物tm-5的质谱图;
[0174]
图12为本发明化合物tm-6的1h-nmr谱图;
[0175]
图13为本发明化合物tm-6的
13
c-nmr谱图;
[0176]
图14为本发明化合物tm-6的质谱图;
[0177]
图15为本发明化合物tm-7的1h-nmr谱图;
[0178]
图16为本发明化合物tm-7的质谱图;
[0179]
图17为本发明化合物tm-8的1h-nmr谱图;
[0180]
图18为本发明化合物tm-8的
13
c-nmr谱图;
[0181]
图19为本发明化合物tm-8的质谱图;
[0182]
图20为本发明化合物tm-9的1h-nmr谱图;
[0183]
图21为本发明化合物tm-9的
13
c-nmr谱图;
[0184]
图22为本发明化合物tm-9的质谱图;
[0185]
图23为本发明化合物tm-10的1h-nmr谱图;
[0186]
图24为本发明化合物tm-10的
13
c-nmr谱图;
[0187]
图25为本发明化合物tm-10的质谱图。
具体实施方式
[0188]
以下结合具体实施例详细地解释本发明,使得本领域技术人员更全面地理解本专利。具体实施例仅用于说明本发明的技术方案,并不以任何方式限定本发明。
[0189]
1(3-(2,6-二氯苯基)-4-羟甲基-5-环丙基异噁唑)中间体的合成路线如下:
[0190][0191]
合成方法如下:
[0192]
1.1 2,6-二氯苯甲醛肟的合成
[0193][0194]
将盐酸羟氨(11g,1eq)和氢氧化钠(6.3g,1.2eq)溶于水中,室温下加入2,6-二氯苯甲醛(25g,0.14mmol,1.2eq)的乙醇(200ml)溶液,90℃下搅拌1小时,冷却至室温后减压蒸馏除去乙醇,抽滤,水洗涤滤饼(2
×
100ml),干燥后得白色固体(2,6-二氯苯甲醛肟)9.46g,收率84%。
[0195]
1.2 2,6-二氯-n-羟基-氯代苯甲醛肟的合成
[0196][0197]
于40℃下,将n-氯代丁二酰亚胺(16.08g,0.12mol,1eq)的dmf(90ml)溶液缓慢滴加到2,6-二氯苯甲醛肟(22.8g,0.12mol,1eq)的dmf(90ml)溶液中,搅拌,tlc监测,反应完全后冷却到室温,将溶液倒入冰水(200ml)中,甲基叔丁基醚萃取3次(3
×
100ml),合并有机相后用水(3
×
100ml)、饱和食盐水(100ml)洗涤。无水硫酸钠干燥酯层后抽滤,减压蒸馏除去有机溶剂得黄色油状粗品,硅胶柱层析法梯度洗脱分离纯化(pe:ea=5:1,v/v)得白色固体(2,6-二氯-n-羟基-氯代苯甲醛肟)26g,收率97%。
[0198]
1.3 3-(2,6-二氯苯基)-5-环丙基异噁唑-4-甲酸甲酯的合成
[0199][0200]
将3-环丙基-3-氧代丙酸甲酯(637.7mg,4.49mmol,1eq)加入到100ml反应瓶,胶塞
密闭,用针管把三乙胺(907.9mg,8.97mmol,2eq)加入到反应瓶内,室温下剧烈搅拌30min,反应液冰浴冷却低于10℃,搅拌下缓慢滴加2,6-二氯-n-羟基-氯代苯甲醛肟(1.0g,4.49mmol,1eq)的乙醇溶液(监测内温<24℃),缓慢升至室温,剧烈搅拌过夜,减压蒸馏除去乙醇,加入乙酸乙酯萃取3次(3
×
100ml),有机层用水(3
×
100ml)、饱和食盐水(100ml)洗涤,无水硫酸钠干燥除去溶剂得油状粗品。硅胶柱层析法梯度洗脱分离纯化(pe:ea=40:1,v/v)得白色固体(3-(2,6-二氯苯基)-5-环丙基异噁唑-4-甲酸甲酯)0.89g,收率55%。
[0201]
1.4 3-(2,6-二氯苯基)-4-羟甲基-5-环丙基异噁唑的合成
[0202][0203]
氮气保护和冰浴条件下将二异丁基氢化铝的甲苯溶液(4.0ml,6.0mmol,2.1eq,1.5m的甲苯溶液)缓慢滴加至3-(2,6-二氯苯基)-5-环丙基异噁唑-4-甲酸甲酯(0.89g,2.8mmol,1eq)的无水thf中,升至室温剧烈搅拌过夜。重新将反应液冷却至0℃,缓慢滴加甲醇(2ml)搅拌10min,反应液倒入50ml冰水混合物中,生成凝胶状悬浮物。硅藻土过滤,用乙酸乙酯(3
×
100ml)萃取三次,合并酯层,用水(3
×
100ml)和饱和食盐水(100ml)洗涤,无水硫酸钠干燥,过滤除去溶剂得白色固体。硅胶柱层析法梯度洗脱分离纯化(pe:ea=20:1,v/v)得到白色固体(3-(2,6-二氯苯基)-4-羟甲基-5-环丙基异噁唑)0.45g,收率56%。
[0204]
2、3-(2,6-二氯苯基)-4-羟甲基-5-异丙基异噁唑的合成
[0205]
2.1 3-(2,6-二氯苯基)-5-异丙基异噁唑-4-甲酸甲酯的合成
[0206][0207]
将异丁酰乙酸甲酯(16.6ml,0.12mol,1eq)加入到100ml反应瓶,胶塞密闭,用针管把三乙胺(33.25ml,0.24mol,2eq)加入到反应瓶内,室温下剧烈搅拌30min,反应液冰浴冷却低于10℃,搅拌下缓慢滴加2,6-二氯-n-羟基-氯代苯甲醛肟(26.6g,0.12mol,1eq)的乙醇溶液(监测内温<24℃),缓慢升至室温,剧烈搅拌过夜,减压蒸馏除去乙醇,加入乙酸乙酯萃取3次(3
×
100ml),有机层用水(3
×
100ml)、饱和食盐水(100ml)洗涤,无水硫酸钠干燥除去溶剂得油状粗品。硅胶柱层析法梯度洗脱分离纯化(pe:ea=40:1,v/v)得白色固体(3-(2,6-二氯苯基)-5-异丙基异噁唑-4-甲酸甲酯)21g,收率56%。
[0208]
2.2 3-(2,6-二氯苯基)-4-羟甲基-5-异丙基异噁唑的合成
[0209][0210]
氮气保护和冰浴条件下将二异丁基氢化铝的甲苯溶液(92ml,0.14mol,2.1eq,1.5μ的甲苯溶液)缓慢滴加至3-(2,6-二氯苯基)-5-异丙基异噁唑-4-甲酸甲酯(20g,0.06mol,1eq)的无水thf中,升至室温剧烈搅拌过夜。重新将反应液冷却至0℃,缓慢滴加甲醇(20ml)搅拌10min,反应液倒入200ml冰水混合物中,生成凝胶状悬浮物。硅藻土过滤,用乙酸乙酯(3
×
100ml)萃取三次,合并酯层,用水(3
×
100ml)和饱和食盐水(100ml)洗涤,无水硫酸钠干燥,过滤除去溶剂得白色固体。硅胶柱层析法梯度洗脱分离纯化(pe:ea=40:1,v/v)得到白色固体(3-(2,6-二氯苯基)-4-羟甲基-5-异丙基异噁唑)18g,收率94%。
[0211]
3 3-(2,6-二氯苯基)-4-羟甲基-5-苯基异噁唑的合成
[0212]
3.1 3-(2,6-二氯苯基)-5-苯基异噁唑-4-甲酸乙酯的合成
[0213][0214]
将苯甲酰乙酸乙酯(5.7ml,50mmol,1eq)加入到100ml反应瓶,胶塞密闭,用针管把三乙胺(13.86ml,100mmol,2eq)加入到反应瓶内,室温下剧烈搅拌30min,反应液冰浴冷却低于10℃,搅拌下缓慢滴加2,6-二氯-n-羟基-氯代苯甲醛肟(10g,50mmol,1eq)的乙醇溶液(监测内温<24℃),缓慢升至室温,剧烈搅拌过夜,减压蒸馏除去乙醇,加入乙酸乙酯萃取3次(3
×
100ml),有机层用水(3
×
100ml)、饱和食盐水(100ml)洗涤,无水硫酸钠干燥除去溶剂得油状粗品。硅胶柱层析法梯度洗脱分离纯化(pe:ea=40:1,v/v)得白色固体(3-(2,6-二氯苯基)-5-苯基异噁唑-4-甲酸甲酯)11.7g,收率65%。
[0215]
3.2 3-(2,6-二氯苯基)-4-羟甲基-5-苯基异噁唑的合成
[0216][0217]
氮气保护和冰浴条件下将二异丁基氢化铝的甲苯溶液(18ml,27mmol,2.1eq,1.5m的甲苯溶液)缓慢滴加至3-(2,6-二氯苯基)-5-苯基异噁唑-4-甲酸甲酯(4.72g,13mmol,1eq)的无水thf中,升至室温剧烈搅拌过夜。重新将反应液冷却至0℃,缓慢滴加甲醇(20ml)搅拌10min,反应液倒入200ml冰水混合物中,生成凝胶状悬浮物。硅藻土过滤,用乙酸乙酯(3
×
100ml)萃取三次,合并酯层,用水(3
×
100ml)和饱和食盐水(100ml)洗涤,无水硫酸钠
干燥,过滤除去溶剂得白色固体。硅胶柱层析法梯度洗脱分离纯化(pe:ea=20:1,v/v)得到白色固体(3-(2,6-二氯苯基)-4-羟甲基-5-苯基异噁唑)9.1g,收率70%。
[0218]
实施例1:化合物tm-1合成路线
[0219][0220]
试验步骤:
[0221]
3-(3-溴苯基)环丁酮
[0222][0223]
在-15℃条件下,向n,n-二甲基甲酰胺(2.1g,24.6mmol)的1,2-二氯乙烷(40ml),溶液中,缓慢滴加三氟甲磺酸酐(11.6g,41.0mmol),在-15℃搅拌30分钟。然后加入3-溴苯乙烯(3.0g,16.4mmol)和2,4,6-三甲基吡啶(2.9g,24.6mmol),室温搅拌过夜。加水猝灭反应,室温搅拌过夜,加入二氯甲烷稀释分离有机相,分别用水和饱和食盐水(200ml)洗涤有机相,无水硫酸镁干燥,抽滤,减压浓缩,硅胶柱层析法梯度洗脱分离纯化(pe:ea=15:1,v/v)得到黄色固体3-(3-溴苯基)环丁酮1.3g,产率35%。
[0224]
3-(3-氧代环丁基)苯甲酸甲酯
[0225][0226]
在室温,一氧化碳气球环境中,将三乙胺(2.2g,21.3mmol)加入3-(3-氧代环丁基)苯甲酸甲酯(1.6g,7.1mmol)和(1,1'-双(二苯基膦基)二茂铁)二氯化钯(520mg,0.7mmol)在甲醇(20ml)和n,n-二甲基甲酰胺(10ml)的混合溶剂中,加热到55℃反应18小时,减压蒸馏除去溶剂并溶于乙酸乙酯中,用水洗涤,有机层用无水硫酸镁干燥,抽滤,减压浓缩,硅胶柱层析法梯度洗脱分离纯化(pe:ea=3:1,v/v)得到黄色油状固体3-(3-氧代环丁基)苯甲酸甲酯1.1g,产率75%。
[0227]
4-(5-溴吡嗪-2-亚甲氧基)-5-环丙基-3-(2,6-二氯苯基)异噁唑
[0228][0229]
在100ml的圆底烧瓶中,放入氢化钠(4.9g,121.6mmol),加入少量石油醚,洗涤氢化钠表面的煤油层,洗涤两次。加入四氢呋喃(30ml),反应瓶置于0℃冰浴下冷却,将2,5-二溴吡嗪(13.1g,55.3mmol)溶于四氢呋喃(10ml),搅拌下滴加到圆底烧瓶中。反应20min后,将1(5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲醇(15.7g,55.3mmol)溶于四氢呋喃(10ml)中,针管缓慢滴加到反应瓶中,升至室温反应12h。反应完毕,将反应液倒入100ml冰水混合物中,然后用乙酸乙酯(3
×
100ml)萃取,合并有机相后用水、饱和食盐水洗涤。无水mgso4进行干燥。硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体6(4-(5-溴吡嗪-2-亚甲氧基)-5-环丙基-3-(2,6-二氯苯基)异噁唑)20.2g,产率为83﹪。
[0230]
3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯
[0231][0232]
100ml三颈烧瓶氮气保护,将6(20.2g,45.8mmol)溶于四氢呋喃(80ml)加入反应瓶,然后将温度降到-78℃,缓慢滴加正丁基锂(1.6m in hexane,30.0ml,48.0mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(20ml)中的3-(3-氧代环丁基)苯甲酸甲酯2(9.0g,43.6mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕后,用饱和氯化铵猝灭反应,用乙酸乙酯萃取,饱和食盐水(200ml)洗涤有机相,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=15:1,v/v)得到黄色固体3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯5.6g,产率为19﹪。
[0233]
3-(3-(5-(5-环丙基-3-(2,6-二氯苯基)异噁唑-4-亚甲氧基)-2-吡嗪)-1-羟基环丁烷)苯甲酸
[0234]
[0235]
将3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯(5.6g,10.1mmol)溶解于thf(10ml)和甲醇(10ml)混合溶剂中,35℃下加入lioh
·
h2o(1.8g,42.6mmol)的水(5ml)溶液,搅拌过夜。减压蒸馏除去有机溶剂,用1n盐酸调节ph至5,加入乙酸乙酯萃取三次,用无水硫酸镁干燥,减压蒸馏除去溶剂,经高压制备液相色谱仪分离纯化(乙腈:水=3:4,v/v)得白色固体tm-1 3-(3-(5-(5-环丙基-3-(2,6-二氯苯基)异噁唑-4-亚甲氧基)-2-吡嗪)-1-羟基环丁烷)苯甲酸3.28g,收率57%。
[0236]
图1所示、1h-nmr(400mhz,dmso-d6):δ12.95(s,1h),8.22-8.21(m,1h),8.12-8.11(m,1h),7.95-7.94(m,1h),7.72(d,j=2hz,1h),7.66-7.49(m,4h),7.48-7.37(m,1h),6.06(s,1h),5.24(s,2h),3.57-3.37(m,1h),2.94-2.89(m,2h),2.55-2.42(m,3h),1.21(d,j=24hz,2h),1.15(d,j=16hz,2h)
[0237]
图2所示,si-ms:m/z[m+h]+:calcd.for c
28h23
cl2n3o5:551.1,found:552.2
[0238]
实施例2:化合物tm-2合成路线
[0239][0240]
合成步骤:
[0241]
3-(3-羟基氮杂环丁烷-1-基)苯甲酸甲酯3b的合成
[0242][0243]
向3-碘苯甲酸甲酯3a(5.0g,19.1mmol)在dmso-d6(70ml)的溶液中加入3-氮杂环环丁烷-3-醇盐酸盐(2.5g.22.9mmol)、cs2co3(15.5g,47.7mmol)、cui(726mg,3.8mmol)和l-脯氨酸(878mg,7.6mmol),然后将该混合物在氩气气氛下在90℃加热18小时。溶液用乙酸乙酯和水稀释,然后将有机层用盐水洗涤三次,减压浓缩,用硅胶柱层析色谱分离纯化(dcm/meoh=10/1,v/v),得到白色固体产物3b(2.7g,68%)。
[0244]
3-(3-氧代氮杂环丁烷-1-基)苯甲酸甲酯3的合成
[0245][0246]
将二甲亚砜(1.6g,20.3mmol)溶于二氯甲烷(30ml)中,在-78℃下,加入草酰氯(1.3g,10.1mmol),并在-78℃搅拌30分钟,然后加入3-(3-羟基氮杂环丁烷-1-基)苯甲酸甲酯(1.4g,6.8mmol)溶于二氯甲烷,在-78℃下缓慢滴加到反应液中,并控制时间在30分钟,然后在-78℃搅拌30分钟,随后加入三乙胺(4.1g,40.5mmol),在-78℃反应1小时,升至室温并在室温下反应2小时。反应液用水稀释,并用乙酸乙酯萃取,有机相用饱和食盐水洗涤,无水硫酸钠干燥,抽滤浓缩,用硅胶柱层析色谱分离纯化(pe/ea=2/1)得到白色固体产物3(0.9g,65%)。
[0247]
3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基氮杂环丁烷-1-基)苯甲酸甲酯
[0248][0249]
100ml三颈烧瓶氮气保护,将6(1.9g,4.4mmol)溶于四氢呋喃(25ml)加入反应瓶,然后将温度降到-78℃,缓慢滴加正丁基锂(2.5m in hexane,2.6ml,6.6mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(5ml)中的3-(3-氧代氮杂环丁烷-1-基)苯甲酸甲酯3(0.9g,4.4mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕后,用饱和氯化铵猝灭反应,用乙酸乙酯萃取,饱和食盐水(200ml)洗涤有机相,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=15:1,v/v)得到黄色固体7 3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯560mg,产率为22﹪
[0250]
3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基氮杂环丁烷-1-基)苯甲酸
[0251][0252]
将3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯(270mg,0.5mmol)溶解于thf(3ml)和甲醇(3ml)混合溶剂中,35℃下加入lioh
·
h2o(60mg,1.5mmol)的水(3ml)溶液,搅拌过夜。减压蒸馏除去有机溶剂,用1n盐酸调节ph至5,加入乙酸乙酯萃取三次,用无水硫酸镁干燥,减压蒸馏除去溶剂,经高压制备液相色谱仪分离纯化(乙腈:水=3:4,v/v)得白色固体3-(3-(5-((5-环丙基-3-(2,6-二
氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基苯甲酸)40mg,收率15%。
[0253]
图3所示、1h-nmr(400mhz,dmso-d6):δ8.22-8.21(m,1h),8.08-8.08(m,1h),7.59-7.51(m,4h),7.27-7.25(m,2h),7.00(s,1h),6.67(s,1h),5.24(s,2h),4.21(d,j=16hz,2h),3.98(d,j=16hz,2h),2.51-2.50(m,1h),1.22-1.18(m,2h),1.15-1.12(m,2h)。
[0254]
图4所示、esi-ms:m/z[m+h]+:calcd.for c
27h22
cl2n4o5:552.1,found:553.1(注,数值在图的右侧)。
[0255]
实施例3:化合物tm-3合成路线
[0256]
3-(3-溴苯基)-1-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)环丁醇
[0257][0258]
100ml三颈烧瓶氮气保护,将4-(5-溴吡嗪-2-亚甲氧基)-5-环丙基-3-(2,6-二氯苯基)异噁唑(1.14g,2.3mmol)溶于无水四氢呋喃(5ml)打入反应瓶,然后将乙醇与液氮加入500ml低温杜瓦瓶使温度降到-78℃,缓慢滴加正丁基锂(1.7ml,2.7mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(10ml)中的3-(3-氧代环丁酮)苯甲酸甲酯(0.56g,2.5mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕将反应液缓慢倒入冰水混合物中,用乙酸乙酯萃取,水(100ml)洗涤酯层,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体3-(3-溴苯基)-(5-(5-环丙基-3-(2,6-二氯苯基)异恶唑-4-基)甲氧基)吡嗪-2-基)环丁烷-1-醇,567mg,产率为42﹪。
[0259]
图5所示、1h-nmr(400mhz,cdcl3):δ8.30(1h,s),8.06(1h,d,j=4hz),7.45(1h,s),7.42(1h,d,j=1.6hz),7.40(1h,s),7.36-7.32(2h,m,j=16hz),7.22-7.18(2h,m,j=16hz),5.23(2h,s),3.35-3.26(1h,m),2.99-2.93(2h,m),2.63-2.57(2h,m),2.36-2.29(1h,m),1.33-1.29(2h,d,j=24hz),1.21-1.16(2h,d,j=16hz);
[0260]
图6所示、
13
c-nmr(100mhz,dmso-d6):173.0,159.6,158.4,150.9,147.1,130.0,129.8,129.3,128.0,127.9,125.3,122.6,110.3,71.0,56.8,44.5,29.8,8.5,7.8。
[0261]
图7所示、esi-ms:m/z[m+2+h]
+
:calcd.forc
27h22
brcl2n3o3:585.0222,found:588.0300
[0262]
实施例4:化合物tm-4合成路线
[0263]
1-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-(3-(甲基磺酰基)苯基)环丁醇
[0264][0265]
向3-(3-溴苯基)-1-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)环丁醇(500mg,0.85mmol)在dmso-d6中的溶液中,加入甲烷亚磺酸钠(130mg,1.28mmol)、cui(50.2mg,0.26mmol)、l-脯氨酸(97.9mg,0.85mmol)和二异丙基乙胺(diea)(109.9mg,0.85mmol)。将该混合物在95℃搅拌过夜,然后用水稀释并用ea萃取。有机相合并,用水洗涤并用na2so4干燥。减压浓缩至干经高压制备液相色谱仪分离纯化(乙腈:水=3:4,v/v)得白色固体1-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-(3-(甲基磺酰基)苯基)环丁醇324mg,收率65%。
[0266]
图8所示、1h-nmr(400mhz,cdcl3):δ8.15(s,1h),7.92(s,1h),7.72-7.65(m,2h),7.42-7.40(m,2h),7.28-7.12(m,2h),5.08(s,2h),3.33-3.29(m,1h),2.93-2.85(m,5h),2.51-2.46(m,2h),2.21-2.18(m,1h),1.15(d,j=16hz,2h),1.05(d,j=16hz,2h)
[0267]
图9所示、esi-ms:m/z[m+h]+:calcd.for c
28h25
cl2n3o5s:585.1,found:586.3
[0268]
实施例5:化合物tm-5合成路线
[0269]
1-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-(3-(苯硫基)苯基)环丁醇
[0270][0271]
在氩气保护下,向3-(3-溴苯基)-1-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)环丁醇(1.0g,1.7mmol)在甲苯中的溶液中加入diea(0.44g,3.41mmol)、苯甲硫醇(0.21g,1.7mmol)、pd2(dba)3(0.34g,0.37mmol)和4,5-双二苯基膦-9,9-二甲基氧杂蒽(0.16g,0.27mmol)。然后将混合物在115℃搅拌4小时。冷却至室温,用水稀释并用ea萃取。有机相合并,用水洗涤并用na2so4干燥。减压浓缩至干经高压制备液相色谱仪分离纯化(乙腈:水=3:4,v/v)得白色固体1-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-(3-(苯硫基)苯基)环丁醇367mg,收率35%。
[0272]
图10所示、1h-nmr(400mhz,cdcl3):δ8.23(s,1h),8.01(s,1h),7.46-7.54(m,3h),7.23-7.33(m,8h),7.14(br d,j=7.6hz,1h),5.28(s,2h),3.31-3.34(m,1h),4.85(h2o),4.58(hdo),3.30(cd3od),2.92-3.02(m,2h),2.41-2.50(m,2h),1.23(s,1h),1.21(br d,j=2.0hz,2h),1.19(br d,j=2.0hz,2h)
[0273]
图11所示、esi-ms:m/z[m+h]+:calcd.for c
33h27
cl2n3o3s:615.1150,found:616.1421。
[0274]
实施例6:化合物tm-6的合成
[0275]
4-(5-溴吡嗪-2-亚甲氧基)-5-异丙基-3-(2,6-二氯苯基)异噁唑的合成
[0276][0277]
在100ml的圆底烧瓶中,放入氢化钠(60%,0.83g,21mmol),加入少量石油醚,洗涤nah表面的煤油层,洗涤两次。加入30ml四氢呋喃,反应瓶置于0℃冰浴下冷却。将2,5-二溴吡嗪(833mg,3.5mmol)溶于10ml四氢呋喃,搅拌下滴加到圆底烧瓶中。反应20min后,将3-(2,6-二氯苯基)-4-羟甲基-5-异丙基异噁唑(1g,3.5mmol)溶于10ml四氢呋喃中,针管缓慢滴加到反应瓶中。升至室温反应12h。反应完毕,将反应液倒入100ml冰水混合物中,然后用乙酸乙酯(3
×
100ml)萃取,合并有机相后用水、饱和食盐水洗涤。无水mgso4进行干燥。硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体4-(5-溴吡嗪-2-亚甲氧基)-5-异丙基-3-(2,6-二氯苯基)异噁唑0.7g,产率为53﹪。
[0278]
3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯的合成
[0279][0280]
100ml三颈烧瓶氮气保护,将4-(5-溴吡嗪-2-亚甲氧基)-5-异丙基-3-(2,6-二氯苯基)异噁唑(1g,2.3mmol)溶于无水四氢呋喃(20ml)打入反应瓶,然后将乙醇与液氮加入500ml低温杜瓦瓶使温度降到-78℃,缓慢滴加正丁基锂(1.7ml,2.7mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(10ml)中的3-(3-氧代环丁酮)苯甲酸甲酯(0.51g,2.5mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕将反应液缓慢倒入冰水混合物中,用乙酸乙酯萃取,水(100ml)洗涤酯层,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯549mg,产率为42﹪。
[0281]
3-(3-(5-(5-异丙基-3-(2,6-二氯苯基)异噁唑-4-亚甲氧基)-2-吡嗪)-1-羟基环丁烷)苯甲酸的合成
[0282][0283]
将3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯(319mg,0.6mmol,1eq)溶解于20ml thf中,35℃下lioh
·
h2o(99mg,2.4mmol,4.2eq)的水(5ml)溶液,搅拌过夜。减压蒸馏除去有机溶剂,用1n盐酸调节
ph至5,加入乙酸乙酯萃取三次,用无水硫酸镁干燥,减压蒸馏除去溶剂,使用高压制备液相色谱仪分离纯化,采用waters xbridge c18柱(150nm*4.6nm*3.5um),流动相为乙腈和水,流速18ml/min,收集梯度为45%-75%的馏分,浓缩除掉大部分乙腈,用冻干机冻干得白色粉末状固体(3-(3-(5-(5-异丙基-3-(2,6-二氯苯基)异噁唑-4-亚甲氧基)-2-吡嗪)-1-羟基环丁烷)苯甲酸)126mg,收率38%。
[0284]
图12所示1h-nmr(400mhz,dmso-d6):δ12.97(s,1h),8.20(d,j=1.3hz,1h),8.09(d,j=1.3hz,1h),7.95(s,1h),7.79-7.77(d,j=7.7hz,1h),7.63-7.61(m,2h),7.55-7.53(m,2h),7.44(t,j=7.7hz,1h),6.09(s,1h),5.19(s,2h),3.61-3.54(m,1h),3.42-3.33(m,1h),2.90(td,j=8.9,2.5hz,2h),2.47-2.42(m,2h),1.37(d,j=7.0hz,6h);
[0285]
图13所示、
13
c-nmr(100mhz,cdcl3):176.8,171.0,159.3,158.4,150.9,145.2,131.2,129.4,128.7,128.4,128.2,128.1,128.0,109.1,71.1,56.8,44.5,29.9,27.0,20.9,1.0;
[0286]
图14所示、esi-ms:m/z[m+h]+:calcd.for c
28h25
cl2n3o5:553.1171,found:554.1211.
[0287]
实施例7:化合物tm-7的合成
[0288]
4-(5-溴吡嗪-2-亚甲氧基)-5-苯基-3-(2,6-二氯苯基)异噁唑的合成
[0289][0290]
在100ml的圆底烧瓶中,放入氢化钠(60%,0.83g,21mmol),加入少量石油醚,洗涤nah表面的煤油层,洗涤两次。加入30ml四氢呋喃,反应瓶置于0℃冰浴下冷却。将2,5-二溴吡嗪(833mg,3.5mmol)溶于10ml四氢呋喃,搅拌下滴加到圆底烧瓶中。反应20min后,将3-(2,6-二氯苯基)-4-羟甲基-5-苯基异噁唑(1.12g,3.5mmol)溶于10ml四氢呋喃中,针管缓慢滴加到反应瓶中。升至室温反应12h。反应完毕,将反应液倒入100ml冰水混合物中,然后用乙酸乙酯(3
×
100ml)萃取,合并有机相后用水、饱和食盐水洗涤。无水mgso4进行干燥。硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体(4-(5-溴吡嗪-2-亚甲氧基)-5-苯基-3-(2,6-二氯苯基)异噁唑),752mg,产率为45﹪。
[0291]
3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯的合成
[0292][0293]
100ml三颈烧瓶氮气保护,将4-(5-溴吡嗪-2-亚甲氧基)-5-苯基-3-(2,6-二氯苯
基)异噁唑(1g,2.1mmol)溶于无水四氢呋喃(20ml)打入反应瓶,然后将乙醇与液氮加入500ml低温杜瓦瓶使温度降到-78℃,滴加正丁基锂溶液(1.1n环己烷溶液,1.75ml,2.8mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(10ml)中的3-(3-氧代环丁酮)苯甲酸甲酯(0.47g,2.3mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕将反应液缓慢倒入冰水混合物中,用乙酸乙酯萃取,水(100ml)洗涤酯层,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯329mg,产率为26﹪。
[0294]
3-(3-(5-(5-苯基-3-(2,6-二氯苯基)异噁唑-4-亚甲氧基)-2-吡嗪)-1-羟基环丁烷)苯甲酸的合成
[0295][0296]
将3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯(118mg,0.2mmol,1eq)溶解于20ml thf中,35℃下lioh
·
h2o(35mg,0.8mmol,4.2eq)的水(5ml)溶液,搅拌过夜。减压蒸馏除去有机溶剂,用1n盐酸调节ph至5,加入乙酸乙酯萃取三次,用无水硫酸镁干燥,减压蒸馏除去溶剂,使用高压制备液相色谱仪分离纯化,采用waters x bridge c18柱(150nm*4.6nm*3.5um),流动相为乙腈和水,流速18ml/min,收集梯度为45%-75%的馏分,浓缩除掉大部分乙腈,用冻干机冻干得白色粉末状固体(3-(3-(5-(5-苯基-3-(2,6-二氯苯基)异噁唑-4-亚甲氧基)-2-吡嗪)-1-羟基环丁烷)苯甲酸)39mg,收率33%。
[0297]
图15所示、1h-nmr(400mhz,dmso-d6):δ12.97(s,1h),8.15(d,j=1.3hz,1h),8.10(d,j=1.3hz,1h),7.99-7.95(m,j=16hz,3h),7.79-7.78(d,j=4hz,1h),7.67-7.65(m,j=8hz,5h),7.61-7.55(m,j=8hz,2h),7.46-7.42(m,1h),6.08(s,1h),5.38(s,2h),5.39-3.33(m,1h),2.94-2.88(m,2h),2.47-2.42(m,2h);
[0298]
图16所示,esi-ms:m/z[m+h]+:calcd.for c
31h23
cl2n3o5:587.1015,found:588.1062
[0299]
实施例8:化合物tm-8的合成
[0300]
[0301]
3-甲基-5-乙烯基苯甲酸甲酯
[0302][0303]
100ml圆底烧瓶,加入3-溴苯甲酸甲酯(1.12g,5mmol),乙烯三氟硼酸钾(820mg,6.12mmol),pdcl2(17.5mg,0.1mmol),pph3(80mg,0.3mmol)和cs2co3(5g,15mmol),然后在n2下加入thf(18ml)和h2o(2ml)。混合物在80℃下搅拌反应22h,冷却至室温后,用水洗涤、无水硫酸镁干燥、抽滤。减压蒸馏,用硅胶柱层析法梯度洗脱分离纯化(pe:ea=60:1,v/v)得到浅粉色油状液体(3-甲基-5-乙烯基苯甲酸甲酯)164mg,产率30%。
[0304]
3-(2,2-二氯-3-氧环丁酮)-5-甲基苯甲酸甲酯
[0305][0306]
氮气保护下,将3-甲基-5-乙烯基苯甲酸甲酯(5.46g,31mmol,1eq)溶解于乙醚(150ml)中。加入锌粉(6g,93mmol,3eq),超声30min后,滴加一种三氯乙酰氯(8.7ml,77.5mmol,2.5eq)的et2o溶液(50ml),继续超声30分钟。混合物加热到35℃。持续超声2.5h,反应完成后冷却至室温,缓慢滴加水(50ml)淬灭。混合物倒入水中搅拌20min后,过滤,再用et2o漂洗。有机层用水(250ml)、饱和碳酸氢钠(250ml)和饱和氯化钠(250ml)洗涤,用无水硫酸镁干燥,过滤后减压蒸馏除去溶剂得黄色油状粗品。硅胶柱层析法梯度洗脱分离纯化(pe:ea=50:1,v/v)得到黄色油状液体3-(2,2-二氯-3-氧环丁酮)-5-甲基苯甲酸甲酯3.56g,产率40%。
[0307]
3-甲基-5-(3-氧环丁基)苯甲酸甲酯
[0308][0309]
混合3-(2,2-二氯-3-氧环丁酮)-5-甲基苯甲酸甲酯(2.79g,9.7mmol,1eq)与锌粉(2.54g,38.8mmol,4eq)溶于60ml乙酸中,室温下搅拌1h。然后在油浴80℃下回流3.5h,反应完毕后冷却至室温。用100ml水稀释溶剂乙酸,用乙醚(3
×
40ml)萃取。合并有机相后依次用饱和碳酸钠溶液(3
×
40ml)、水(100ml)、饱和食盐水(100ml)洗涤。用一定量的无水mgso4进行干燥。硅胶柱层析法梯度洗脱分离纯化(pe:ea=50:1,v/v)得到化合物(3-(3-氧代环丁酮)苯甲酸甲酯)1.38g,产率65%。
[0310]
3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯的合成
[0311][0312]
100ml三颈烧瓶氮气保护,将4-(5-溴吡嗪-2-亚甲氧基)-5-环丙基-3-(2,6-二氯苯基)异噁唑(1.02g,2.3mmol)溶于无水四氢呋喃(20ml)打入反应瓶,然后将乙醇与液氮加入500ml低温杜瓦瓶使温度降到-78℃,缓慢滴加正丁基锂(1.7ml,2.7mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(10ml)中的3-甲基-5-(3-氧环丁基)苯甲酸甲酯(0.55g,2.5mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕将反应液缓慢倒入冰水混合物中,用乙酸乙酯萃取,水(100ml)洗涤酯层,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯,产率为47﹪。
[0313]
3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸
[0314][0315]
将3-(3-(5-((3-(2,6-二氯苯基)-5-环丙基异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)苯甲酸甲酯(116mg,0.2mmol,1eq)溶解于20ml thf中,35℃下lioh
·
h2o(35mg,0.8mmol,4.2eq)的水(5ml)溶液,搅拌过夜。减压蒸馏除去有机溶剂,用1n盐酸调节ph至5,加入乙酸乙酯萃取三次,用无水硫酸镁干燥,减压蒸馏除去溶剂,使用高压制备液相色谱仪分离纯化,采用waters xbridge c18柱(150nm*4.6nm*3.5um),流动相为乙腈和水,流速18ml/min,收集梯度为45%-75%的馏分,浓缩除掉大部分乙腈,用冻干机冻干得白色粉末状固体3-(3-(5-((5-环丙基-3-(2,6-二氯苯基)异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸,37mg,收率33%。
[0316]
图17所示、1h-nmr(400mhz,dmso-d6):δ12.90(1h,s),8.21(1h,d,j=1.2hz),8.11(1h,d,j=1.2hz),7.74(1h,s),7.62-7.60(3h,m),7.56-7.52(1h,m),7.37(1h,s),6.06(1h,s),5.24(2h,s),3.38-3.29(1h,m),2.92-2.87(2h,m),2.59-2.53(1h,m),2.46-2.41(2h,m),2.35(3h,s),1.24-1.18(2h,m),1.16-1.14(2h,m);
[0317]
图18所示、
13
c-nmr(400mhz,dmso-d6):δ176.9,168.9,168.0,159.3,157.9,153.5,146.2,135.1,133.4,133.0,128.9,127.6,125.1,109.9,70.9,56.3,45.5,29.8,26.5,21.3,21.1;
[0318]
图19所示、esi-ms:m/z[m+h]+:calcd.for c
29h25
cl2n3o5:565.1171,found:
566.1234
[0319]
实施例9:化合物tm-9的合成
[0320]
3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯的合成
[0321][0322]
100ml三颈烧瓶氮气保护,将4-(5-溴吡嗪-2-亚甲氧基)-5-异丙基-3-(2,6-二氯苯基)异噁唑(1.02g,2.3mmol)溶于无水四氢呋喃(20ml)打入反应瓶,然后将乙醇与液氮加入500ml低温杜瓦瓶使温度降到-78℃,缓慢滴加正丁基锂(1.7ml,2.7mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(10ml)中的3-甲基-5-(3-氧环丁基)苯甲酸甲酯(0.55g,2.5mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕将反应液缓慢倒入冰水混合物中,用乙酸乙酯萃取,水(100ml)洗涤酯层,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v,pe是石油醚,ea是乙酸乙酯)得到白色固体(3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯)643mg,产率为48﹪。
[0323]
3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸的合成
[0324]
将3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯(117mg,0.2mmol,1eq)溶解于20ml thf中,35℃下lioh
·
h2o(35mg,0.8mmol,4.2eq)的水(5ml)溶液,搅拌过夜。减压蒸馏除去有机溶剂,用1n盐酸调节ph至5,加入乙酸乙酯萃取三次,用无水硫酸镁干燥,减压蒸馏除去溶剂,使用高压制备液相色谱仪分离纯化,采用waters x bridge c18柱(150nm*4.6nm*3.5um),流动相为乙腈和水,流速18ml/min,收集梯度为45%-75%的馏分,浓缩除掉大部分乙腈,用冻干机冻干得白色粉末状固体(3-(3-(5-((3-(2,6-二氯苯基)-5-异丙基异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸),46mg,收率40%。
[0325]
图20所示、1h-nmr(400mhz,dmso-d6):δ12.88(1h,brs),8.20(1h,d,j=1.2hz),8.08(1h,d,j=1.2hz),7.73(1h,s),7.63-7.60(3h,m),7.56-7.52(1h,s),7.37(1h,s),6.06(1h,brs),5.19(2h,s),3.61-3.54(1h,m),2.91-2.85(2h,m),2.45-2.40(2h,m),2.35(3h,s),1.38-1.36(6h,d,j=8hz);
[0326]
图21所示、
13
c-nmr(100mhz,dmso-d6):176.9,168.0,159.3,157.9,153.5,146.2,135.1,133.4,133.0,128.9,127.6,125.1,109.9,70.9,56.3,45.5,29.8,26.5,21.3,21.1;
[0327]
图22所示、esi-ms:m/z[m+h]+:calcd.for c
29h27
cl2n3o5:567.1328,found:568.1412
[0328]
实施例10:化合物tm-10的合成
[0329]
3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯的合成
[0330][0331]
100ml三颈烧瓶氮气保护,将4-(5-溴吡嗪-2-亚甲氧基)-5-苯基-3-(2,6-二氯苯基)异噁唑(1.03g,2.3mmol)溶于无水四氢呋喃(20ml)打入反应瓶,然后将乙醇与液氮加入500ml低温杜瓦瓶使温度降到-78℃,缓慢滴加正丁基锂(1.7ml,2.7mmol),搅拌10min后,缓慢滴加溶于四氢呋喃(10ml)中的3-甲基-5-(3-氧环丁基)苯甲酸甲酯(0.55g,2.5mmol)溶液,-78℃反应2h后升至室温反应过夜。反应完毕将反应液缓慢倒入冰水混合物中,用乙酸乙酯萃取,水(100ml)洗涤酯层,无水硫酸镁干燥,抽滤,减压蒸馏除去有机溶剂,硅胶柱层析法梯度洗脱分离纯化(pe:ea=10:1,v/v)得到白色固体(3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异噁唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯)496mg,产率为35﹪。
[0332]
3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸的合成
[0333][0334]
将3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸甲酯(123mg,0.2mmol,1eq)溶解于20ml thf中,35℃下lioh
·
h2o(35mg,0.8mmol,4.2eq)的水(5ml)溶液,搅拌过夜。减压蒸馏除去有机溶剂,用1n盐酸调节ph至5,加入乙酸乙酯萃取三次,用无水硫酸镁干燥,减压蒸馏除去溶剂,使用高压制备液相色谱仪分离纯化,采用waters xbridge c18柱(150nm*4.6nm*3.5um),流动相为乙腈和水,流速18ml/min,收集梯度为45%-75%的馏分,浓缩除掉大部分乙腈,用冻干机冻干得白色粉末状固体(3-(3-(5-((3-(2,6-二氯苯基)-5-苯基异恶唑-4-基)甲氧基)吡嗪-2-基)-3-羟基环丁基)-5-甲基苯甲酸),37mg,收率31%。
[0335]
图23所示、1h-nmr(400mhz,dmso-d6):δ12.89(1h,s),8.14(1h,d,j=1.6hz),8.09(1h,d,j=1.6hz),7.99-7.96(3h,m),7.73(1h,s),7.67-7.65(5h,m),7.61-7.57(2h,m),7.37(1h,s),6.06(1h,s),5.38(2h,s),3.33-3.28(1h,m),2.91-2.86(2h,m),2.45-2.40(2h,m),2.35(3h,s);
[0336]
图24所示、
13
c-nmr(100mhz,dmso-d6):168.4,168.0,160.4,157.8,153.7,146.2,135.2,133.3,131.6,130.0,129.0,127.7,127.2,125.1,111.5,70.9,57.0,45.5,29.8,21.3;
[0337]
图25所示、esi-ms:m/z[m+h]+:calcd.for c
32h25
cl2n3o5:601.1171,found:602.1249。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1