依托度酸原料药中一个微量杂质的分离制备方法与流程
本发明属于药物杂质研究
技术领域:
,具体涉及高速逆流色谱法从依托度酸原料药中分离制备一个微量杂质的方法。
背景技术:
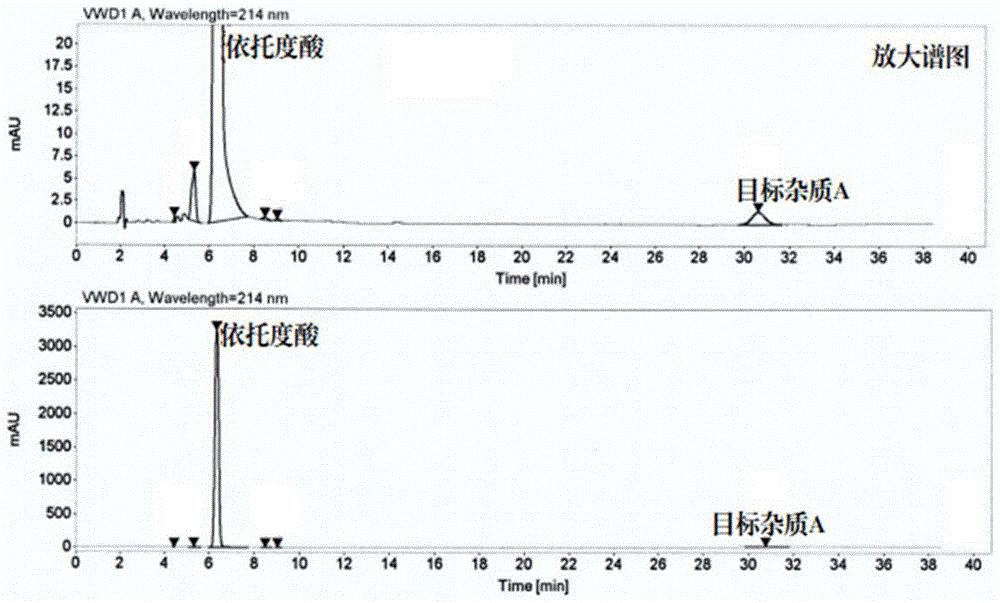
:药物生产过程中常需加入试剂、溶剂或催化剂,由于溶解度、吸附、吸留、共沉淀、混晶生成等原因,不可能完全除去,使产品中存在有关杂质。如使用酸性或碱性试剂处理后,可能使产品中带有酸性或碱性杂质;用有机溶剂提取或精制后,在产品中就可能有残留有机溶剂。中国药典中规定必须检查药物在生产过程中引入的有害有机溶剂(如苯、氯仿、1,4-二氧六环、二氯甲烷、吡啶等)的残留量。药物的杂质与药品安全性的关系是一个受很多因素影响的复杂的关系,通常药物中的杂质大多具有潜在的生物活性,有的甚至与药物相互作用从而影响药物的效能和安全性,严重的可能产生毒性作用。为达到进一步评价药物毒、副作用及避免不良反应的目的,对药物的杂质进行分离、鉴定是一项关键性的技术。依托度酸(etodolac)又称乙哚酸、吲哚吡喃乙酸、罗丁等,商品名为lodine,是新一代的cox-2高选择性非甾体抗炎药,其作用效果与阿司匹林相似,是在炎症部位通过阻断环氧合酶的活性,从而有选择性地抑制前列腺素的生物合成。对胃pge2的抑制轻微、短暂,因此胃肠道不良反应较小。口服吸收迅速。依托度酸最早是由美国的ahp的子公司开发,于1985年在英国成功上市,后在法国、美国、德国、日本等国相继上市。依托度酸在生产过程中会形成微量杂质,其中包括2-(7-乙基-1h-吲哚-2-基)乙酸乙酯2-(1,8-二乙基-1,3,4,9-四氢吡喃[3,4-b]吲哚-1-基(以下简称杂质a),其分子式为c29h34n2o3,化学结构如式i所示。由于杂质a在依托度酸中的含量极低(<0.5%),采用传统的分离方法(如萃取、重结晶、蒸馏和传统柱色谱)很难将其分离,而制备型高效液相色谱由于使用昂贵的色谱级试剂以及c18制备色谱柱,使用成本高、分离纯化时间长。与传统的液-固色谱方法相比,近年来逐渐在分离科学领域绽露头脚的一项液-液分配色谱技术---高速逆流色谱(high-speedcountercurrentchromatography,hsccc),由于不使用固相载体而显示出了与众不同的优势,作为现有液-固制备色谱方法的有效补充,已成为颇具前景的一项制备型色谱技术。逆流色谱利用两互不相溶的溶剂体系在高速旋转的螺旋管内建立起一种特殊的单向性流体动力学平衡,使物质的分离依据其在两相中分配系数的不同而实现。由于不采用固体支撑体,该技术完全排除了因不可逆吸附而引起的样品污染、变性、失活等,不仅简化了样品的预处理工作,且可以通过多种洗脱方式对滞留于色谱柱中的样品予以完全回收。此外,该方法操作成本低廉,制备量易于放大(根据需要一次上样量可以从几毫克到十几克不等)。技术实现要素:本发明要解决的技术问题是提供一种从依托度酸中分离制备微量杂质a的方法;以便于对依托度酸系列产品进行质量控制。为了解决上述技术问题,本发明提供一种依托度酸原料药中一个微量杂质(微量杂质a)的分离制备方法,微量杂质的分子式为c29h34n2o3,采用梯度洗脱的高速逆流色谱法对依托度酸原料药中的微量杂质进行分离制备;逆流色谱法的溶剂体系为正己烷:乙酸乙酯:乙醇:水;洗脱模式为梯度洗脱;通过紫外检测器的在线监测,收集、合并目标流分并旋转蒸发至干,得微量杂质。作为本发明的依托度酸原料药中一个微量杂质的分离制备方法的改进,包括以下步骤:1)、配制两相溶剂:溶剂体系①:将正己烷:乙酸乙酯:乙醇:水=3:(2±0.1):(3±0.1):(2±0.1)的体积比,配制后倒入分液器中,形成两种互不相溶的两相溶剂,分别得溶剂体系①的上相与下相;溶剂体系②:将正己烷:乙酸乙酯:乙醇:水=2:(1±0.1):(2±0.1):(1±0.1)的体积比,配制后倒入分液器中,形成两种互不相溶的两相溶剂,分别得溶剂体系②的上相与下相;2)、将溶剂体系①的上相与下相按照1:1的体积比混合,得混合溶剂①;将依托度酸原料药溶于所述混合溶剂①中,得到依托度酸原料药溶液,所述依托度酸原料药与混合溶剂①的料液比为0.4~0.6g/6~10ml;3)、将溶剂体系①的上相注入逆流色谱仪分离柱内作为固定相,将步骤2)所得的依托度酸原料药溶液注入逆流色谱仪的进样环中,开启逆流色谱仪,流动相的流速为4.0~6.0ml/min;采用梯度洗脱模式:0~100min注入溶剂体系①的下相作流动相(100%);100~180min注入溶剂体系②的下相作流动相进行线性梯度洗脱,溶剂体系②的下相为0~100%(线性梯度洗脱);即,使得溶剂体系②的下相从100min时的0%含量均匀的变化到180min时的100%含量;180min起以溶剂体系②的下相作流动相(100%)进行洗脱;直至到330min时结束洗脱,收集保留时间为290~305min的组分,旋转蒸发(温度为45±5℃,至旋干)后得到微量杂质(为纯化的杂质)。作为本发明的依托度酸原料药中一个微量杂质的分离制备方法的进一步改进:溶剂体系①:正己烷:乙酸乙酯:乙醇:水=3:2:3:2的体积比;溶剂体系②:正己烷:乙酸乙酯:乙醇:水=2:1:2:1的体积比。作为本发明的依托度酸原料药中一个微量杂质的分离制备方法的进一步改进:依托度酸原料药与混合溶剂①的料液比为500mg/8ml。作为本发明的依托度酸原料药中一个微量杂质的分离制备方法的进一步改进:所述流动相的流速为5.0ml/min。备注说明:注入流动相后,杂质便在逆流色谱仪内进行连续的分配。使用在线混合器的梯度控制器执行梯度运行,通过紫外检测器的在线监测,收集、合并流出物(收集保留时间为290~305min的组分)。本发明所述的依托度酸原料药中的杂质为微量杂质,其分子量为458.59da,分子式为c29h34n2o3,化学结构式如上述(i)所示。高速逆流色谱目前尚未用于依托度酸原料药中微量杂质(微量杂质a)的分离制备的原因是:未发现能够分离微量杂质的逆流色谱两相溶剂体系及洗脱模式,即,本发明在发明过程中克服的技术难题是:建立了以正己烷:乙酸乙酯:乙醇:水溶液为两相溶剂体系,并采用梯度洗脱模式,实现了逆流色谱中依托度酸原料药与微量杂质的基线分离。本发明采用的逆流色谱方法选择的是梯度洗脱模式,梯度洗脱模式可以有效改善单一溶剂体系在化合物分离中因极性窗口狭小难以达到理想分离效果的缺陷。梯度洗脱普遍用来分离极性差异偏大的目标物质,其优势是有效减少溶剂消耗、缩短分离时间、柱的选择性和峰容量都将大大提高,这对复杂样品非常有用。逆流色谱分离量大、样品无损失、回收率高,能直接分离大量依托度酸原料药,分离得到的杂质纯度一般可达到98%以上。本发明为生产依托度酸原料药系列产品的质量研究及杂质定量控制提供了便捷的杂质对照品制备方法,可以为申报临床用药制定杂质限量提供可靠依据。综上所述,为了获得高纯度、高回收率的杂质(分子式为c29h34n2o3),本发明提供一种有效、经济的方法对依托度酸原料药中的杂质进行高效分离纯化制备,以便于对依托度酸系列产品进行质量控制。即,本发明提供了采用高速逆流色谱梯度洗脱模式从依托度酸原料药中分离制备一个微量杂质(2-(7-乙基-1h-吲哚-2-基)乙酸乙酯2-(1,8-二乙基-1,3,4,9-四氢吡喃[3,4-b]吲哚-1-基),简称杂质a)的方法。附图说明下面结合附图对本发明的具体实施方式作进一步详细说明。图1为实施例1中依托度酸原料药的高效液相分析图;图2为实施例1中采用梯度洗脱模式分离微量杂质a的逆流色谱图;图3为实施例1中分离纯化的杂质a的高效液相色谱分析图;图4为实施例1中分离纯化的杂质a的核磁共振氢谱图;图5为实施例1中分离纯化的杂质a的核磁共振碳谱图;图6为实施例1中分离纯化的杂质a的核磁共振dept135谱图;图7为实施例1中分离纯化的杂质a的二维核磁共振碳谱图(cosy);图8为实施例1中分离纯化的杂质a的二维核磁共振碳谱图(hsqc);图9为实施例1中分离纯化的杂质a的二维核磁共振碳谱图(hmbc)。具体实施方式下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:以下案例所用的依托度酸原料药,经过分析型高效液相色谱面积归一化法检测,加巴喷丁原料药的含量为99.65%,微量杂质a的含量为0.15%。依托度酸原料药及杂质a的高效液相色谱分析均依据以下高效液相色谱法进行:色谱柱:c18柱,4.6mm×25cm×5um或等同的色谱柱;检测波长:uv214nm;流速:1.0ml/min;进样量:20ul;柱温:30℃;运行时间:40min;流动相:称取5g磷酸,加入300ml水溶解,用0.45um滤膜过滤,再加入700ml乙腈,混合脱气。实施例1、一种从依托度酸原料药中分离制备杂质a的方法,用逆流色谱仪从依托度酸原料药中分离杂质a,依次进行以下步骤:1)、配制两相溶剂:溶剂体系①:将正己烷:乙酸乙酯:乙醇:水=3:2:3:2的体积比,配制后倒入分液器中,形成两种互不相溶的两相溶剂,分别得溶剂体系①的上相与下相;溶剂体系②:将正己烷:乙酸乙酯:乙醇:水=2:1:2:1的体积比,配制后倒入分液器中,形成两种互不相溶的两相溶剂,分别得溶剂体系②的上相与下相;2)、4ml溶剂体系①上相和4ml溶剂体系①下相混合,得混合溶剂①;将500mg依托度酸原料药溶于所述混合溶剂①中,得到依托度酸原料药溶液(样品溶液),即,依托度酸原料药与混合溶剂①的料液比为0.5g/8ml;3)、逆流色谱柱内径2.6mm,柱体积为310ml。以溶剂体系①的上相作为固定相、下相为初始流动相,待逆流色谱仪分离柱内中注满固定相后,将样品溶液注入逆流色谱仪的进样环中,开启逆流色谱仪,转速为1200转,流动相的流速为5.0ml/min。0~100min注入溶剂体系①的下相作流动相(100%),100~180min注入溶剂体系②的下相作流动相(0~100%,线性梯度洗脱)),180min起以溶剂体系②的下相作流动相(100%)进行洗脱;直至到330min时结束洗脱。通过在线的紫外检测器监测流分,检测波长214nm,收集、合并流出物(收集保留时间为290~305min的组分),旋转蒸发(温度为45℃,至旋干),得到0.7mg微量杂质a,其纯度为99.2%,回收率为92.6%。回收率的计算公式为:(ma×pa)/(mp×ca)×100%,其中ma为纯化获得的杂质a的质量,pa为纯化获得的杂质a的纯度,mp为一次进样依托度酸原料药的质量,ca为依托度酸原料药中杂质a的含量。分离杂质a的高速逆流色谱图,如图2所述;分离得到的杂质a的高效液相分析图,如图3所述。分离获得的杂质a的nmr谱图(图4~图9)示出18组质子峰,对应分子中的34个质子。各质子峰的归属如下:0.836(3h)的质子依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-27位的甲基质子;1.369(6h)的质子依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-25、c-29位上的甲基质子;1.983-2.228(2h)的质子依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-26位上的亚甲基质子;2.740-2.867(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-9位上的亚甲基质子;2.843(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-24位上的亚甲基质子;2.883(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-28位上的亚甲基质子;2.997(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-12位上的亚甲基质子;3.103(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-15位上的亚甲基质子;3.935-4.086(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-10位上的亚甲基质子;4.422(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-14位上的亚甲基质子;6.918(1h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-17位上的次甲基质子;7.036(1h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-21位上的次甲基质子;7.051(1h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-2位上的次甲基质子;7.083-7.124(2h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-3、c-22位上的次甲基质子;7.398(1h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-4位上的次甲基质子;7.477(1h)依据化学位移值及相关谱(1h-1hcosy、hsqc、hmbc),归属为c-23位上的次甲基质子;7.866和9.859(2h)依据化学位移值,归属为胺上的活泼氢原子;h和c-1至c-29的化学位移归属如表1、表2所示,,与杂质a的化学结构一致。表1杂质a的1h-nmr图谱数据解析列表表2杂质a的13c-nmr图谱数据解析列表实施例2-1、一种从依托度酸原料药中分离制备杂质a的方法:将实施例1步骤2)中流动相的注入流速由5.0ml/min改成6.0ml/min,其余等同于实施例1。最终所得的杂质a,其纯度为96.4%,回收率为87.7%。实施例2-2、一种从依托度酸原料药中分离制备杂质a的方法:将实施例1步骤2)中流动相的注入流速由5.0ml/min改成4.0ml/min,其余等同于实施例1。最终所得的杂质a,其纯度为96.2%,回收率为85.2%。实施例3、一种从依托度酸中分离制备杂质a的方法:将实施例1步骤2)中依托度酸原料药的用量由500mg改成600mg,且,将流动相的注入流速由5.0ml/min改成6.0ml/min,其余等同于实施例1。最终所得的杂质a,其纯度为93.1%,回收率为83.4%。对比例1-1、将实施例1中的溶剂体系①正己烷、乙酸乙酯、乙醇、水(体积比为3:2:3:2)改成正己烷、乙酸乙酯、乙醇、水(体积比为5:4:5:4),其余同实施例1。对比例1-2、将实施例1中的溶剂体系①正己烷、乙酸乙酯、乙醇、水(体积比为3:2:3:2)改成正己烷、乙酸乙酯、乙醇、水(体积比为1:2:1:2),其余同实施例1。对比例2、将实施例1中的溶剂体系②正己烷、乙酸乙酯、乙醇、水(体积比为2:1:2:1)改成正己烷、乙酸乙酯、乙醇、水(体积比为3:1:3:1),其余同实施例1。对比例3、将实施例1中的溶剂体系②正己烷、乙酸乙酯、乙醇、水(体积比为2:1:2:1)改成正己烷、乙酸乙酯、乙醇、水(体积比为1:1:1:1),其余同实施例1。对比例4-1、将实施例1中的梯度洗脱模式改为仅仅采用溶剂体系①进行等度洗脱,即,具体为:0~500min均注入溶剂体系①的下相作流动相;其余同实施例1。对比例4-2、将实施例1中的梯度洗脱模式改为仅仅采用溶剂体系②进行等度洗脱,即,具体为:0~500min均注入溶剂体系②的下相作流动相;样品溶液也改为溶剂体系②的上相和下相=1:1的混合液进行溶解;其余同实施例1。上述所有对比例所得的杂质a的纯度及回收率如表3所述。表3杂质a纯度回收率对比例1-183.2%80.7%对比例1-279.3%72.7%对比例287.4%84.1%对比例373.5%70.6%对比例4-179.6%50.2%对比例4-268.3%60.9%最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1