一种苯并呋喃类衍生物及其制备方法和应用与流程
1.本发明属于医药领域,涉及一种苯并呋喃类衍生物及其制备方法和应用。
背景技术:
2.目前,恶性肿瘤是世界上病死率最高的疾病之一,仅次于心脑血管疾病,因此抗癌药物的研发具有非常重要的临床和社会意义。有研究表明,蛋白激酶信号通路在癌症形成过程中起着重要作用。磷脂酰肌醇3激酶/蛋白激酶b/哺乳动物雷帕霉素靶蛋白(pi3k/akt/mtor)信号传导通路是哺乳动物细胞内重要的信号传导通路之一,它通过影响其下游多种效应分子的活化状态,发挥抑制细胞凋亡、促进增殖的作用,在癌症患者中可见该通路被过度激活,是人类癌症中最常改变的途径之一。研究表明,pi3k/akt/mtor信号传导通路通过调控促癌基因及各种重要蛋白转录、翻译及表达,抑制癌细胞自噬性死亡,发挥了促癌细胞增殖、抗凋亡、促血管生成、转移及耐药等作用。
3.一些抗肿瘤药物通过抑制akt信号通路的传导来发挥治疗作用,一些中草药例如姜黄素,黄岑苷等被证明是通过抑制pi3k及akt磷酸化、促进凋亡相关蛋白表达来抑制癌症细胞增殖。但目前大部分akt蛋白激酶抑制剂用药成本高,吸收效果差,容易产生抗药性。因此开发一种新型的毒副作用小,易吸收的靶向异常调节途径并最终导致疾病的激酶抑制剂,对于医药界具有巨大的伦理和商业上的利益。
4.本发明创新性的合成了一种苯并呋喃类衍生物,对akt蛋白激酶活性有良好的抑制作用,同时可以抑制异常肿瘤细胞生长,抗肿瘤活性显著,且靶向作用强,毒副作用小,以期得到可以用于制备akt蛋白激酶抑制剂的全新化合物,现有技术并未见到相关结构的报道。
技术实现要素:
5.针对上述问题,本发明的目的是提供一种苯并呋喃类衍生物的制备方法和应用,此类化合物能够有效抑制akt蛋白激酶活性,抗肿瘤活性强,对一些过度增生性疾病如癌症具有良好的治疗效果,可用于制备抗肿瘤的药物。为实现上述目的,本发明采用以下技术方案。
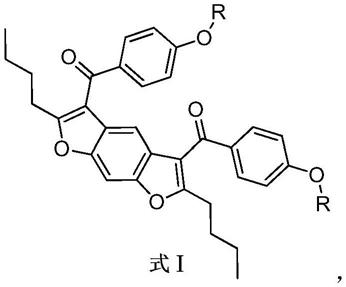
6.一种苯并呋喃类衍生物,其结构通式i如下:
7.8.其中r=
9.本发明另一目的为提供苯并呋喃类衍生物式i的合成路线为:
[0010][0011]
进一步地,上述合成路线中各步骤的合成方法如下:
[0012]
1)在合适的溶剂中,以2,3
‑
双(溴甲基)苯
‑
1,4
‑
二醇与三苯基膦为原料反应得到2,3
‑
双((溴代三苯基
‑
λ5‑
磷烷基)甲基)苯
‑
1,4
‑
二醇;
[0013]
2)以2,3
‑
双((溴代三苯基
‑
λ5‑
磷烷基)甲基)苯
‑
1,4
‑
二醇为原料,反应得到2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃;
[0014]
3)以2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃为原料反应得到(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
甲氧基苯基)甲酮);
[0015]
4)以(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
甲氧基苯基)甲酮)为原料反应得到(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮);
[0016]
5)在碱性条件下,以(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮)和相应的氯化物为原料反应得到对应的最终产物。
[0017]
部分化合物的1h
‑
nmr(400mhz)和
13
c
‑
nmr(125mhz)如下:
[0018]
中间体(结构2所示化合物):1h
‑
nmr(400mhz,cdcl3)δ:2.60(d,4h),6.49(s,2h),7.15(m,12h),7.44
‑
7.47(m,18h),9.68(s,2h).
13
c
‑
nmr(125mhz,cdcl3)δ:29.58,118.00,
123.17,126.49,131.37,132.66,134.54,147.30.
[0019]
中间体(结构3所示化合物):1h
‑
nmr(400mhz,cdcl3)δ:0.90(t,6h),1.30(m,4h),1.59(m,4h),2.87(t,4h),6.72(s,2h),7.64(s,1h),7.97(s,1h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,22.40,29.29,31.49,94.62,103.98,116.02,122.59,149.73,157.55.
[0020]
中间体(结构4所示化合物):1h
‑
nmr(400mhz,cdcl3)δ:0.90(t,6h),1.30(m,4h),1.59(m,4h),3.18(t,4h),3.79(s,6h),7.10(d,4h),7.54(s,1h),7.73
‑
7.97(m,5h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,22.40,27.95,29.26,56.08,96.39,114.40,117.53,118.58,118.69,129.77,134.06,159.33,164.47,172.69,193.50.
[0021]
中间体(结构5所示化合物):1h
‑
nmr(400mhz,cdcl3)δ:0.90(t,6h),1.30(m,4h),1.59(m,4h),2.91(t,4h),5.00(s,2h),6.88(d,4h),7.68(d,4h),7.78(s,1h),8.04(s,1h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,22.40,27.95,29.26,96.39,115.77,117.53,118.58,118.69,129.41,133.29,159.33,163.59,172.69,193.50.
[0022]
化合物1(结构6所示化合物):1h
‑
nmr(400mhz,cdcl3)δ:0.87
‑
0.94(m,18h),1.21
‑
1.34(m,12h),1.35
‑
1.48(m,8h),1.59(m,4h),1.83(m,4h),2.38(t,8h),2.48(t,4h),2.91(t,4h),4.05(t,4h),7.12(d,4h),7.76(s,2h),7.84(d,4h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,20.75,22.40,27.95,28.13,29.25,29.26,51.76,56.45,67.16,96.39,115.25,117.53,118.58,118.69,129.90,134.17,159.33,165.76,172.69,193.50.
[0023]
本发明所述的一种苯并呋喃类衍生物能够有效抑制akt蛋白激酶活性,抗肿瘤活性强,对一些过度增生性疾病如癌症具有良好的治疗效果。说明本发明所述的苯并呋喃类衍生物在作为akt蛋白酶抑制剂治疗或预防肿瘤中具有积极意义,可以进行更加深入的研究。
[0024]
所述的预防或治疗肿瘤的药物或其药学上可接受的盐或溶剂化物在研制针对akt蛋白激酶抑制剂治疗药物中的应用,特别是提供了一种苯并呋喃类衍生物在治疗或预防肿瘤的应用。
[0025]
与现有技术比,本发明的有益效果如下:
[0026]
本发明所述的一种苯并呋喃类衍生物的肿瘤抑制活性强,在多个肿瘤细胞株中均显示出良好抑制作用;还有很好的akt蛋白激酶抑制作用,为研制针对akt蛋白激酶通路癌症的治疗药物具有很好的开发前景。
[0027]
显然,根据本发明的上述内容,按照本领域的普通技术知识和手段,在不脱离本发明上述基本技术思想前提下,还可以做出其他多种形式的修改、替换或变更。
附图说明
[0028]
图1:本发明所得苯并呋喃类衍生物对akt酶活性的影响
[0029]
图2:本发明所得苯并呋喃类衍生物对肿瘤细胞增殖的影响
[0030]
图3:本发明所得苯并呋喃类衍生物对pi3k/akt/mtor信号通路蛋白表达的影响(western blot法)
具体实施方式
[0031]
下列合成实例、生物测试结果可用来进一步说明本发明,但不意味着限制本发明。
[0032]
合成实例
[0033]
实施例1、化合物1的制备
[0034][0035]
(1)中间体2,3
‑
双((溴代三苯基
‑
λ5‑
磷烷基)甲基)苯
‑
1,4
‑
二醇(结构式2所示化合物)的合成:
[0036]
将2,3
‑
双(溴甲基)苯
‑
1,4
‑
二醇(结构式1所示化合物)(100mmol)和三苯基膦(210mmol)在160.0ml氯仿中回流加热0.5小时。然后使反应混合物冷却,滤出形成的白色沉淀。将滤液真空蒸发至干,将得到的粗产品溶于50.0ml甲苯中打浆,过滤,用甲苯洗涤,合并形成的固体并在50℃下真空干燥,得66.46g类白色固体,产率为81%。
[0037]1h
‑
nmr(400mhz,cdcl3)δ:2.60(d,4h),6.49(s,2h),7.15(m,12h),7.44
‑
7.47(m,18h),9.68(s,2h).
13
c
‑
nmr(125mhz,cdcl3)δ:29.58,118.00,123.17,126.49,131.37,132.66,134.54,147.30.
[0038]
(2)中间体2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃(结构式3所示化合物)的合成
[0039]
在搅拌下将戊酰氯(110mmol)缓慢加入到2,3
‑
双((溴代三苯基
‑
λ5‑
磷烷基)甲基)苯
‑
1,4
‑
二醇(结构式2所示化合物)(50mmol)和吡啶(200mmol)的氯仿(100ml)溶液中。然后将混合物加热回流2小时。加入甲苯(280.0ml),蒸馏出140.0ml溶剂。然后加入三乙胺(225mmol),将混合物加热回流3小时。将其冷却,过滤出形成的三苯基膦氧化物,用乙酸乙酯洗涤固体。将滤液真空浓缩至干。将得到的油状物溶解在乙腈中,向溶液中滴加入正戊烷析晶,过滤,真空50℃干燥,得10.54g棕黄色固体,产率78%。
[0040]1h
‑
nmr(400mhz,cdcl3)δ:0.90(t,6h),1.30(m,4h),1.59(m,4h),2.87(t,4h),6.72(s,2h),7.64(s,1h),7.97(s,1h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,22.40,29.29,31.49,
94.62,103.98,116.02,122.59,149.73,157.55.lc
‑
ms(esi,pos,ion)m/z:271.15[m+h].
[0041]
(3)中间体(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
甲氧基苯基)甲酮)(结构式4所示化合物)的合成
[0042]
在0
‑
5℃下,将四氯化锡(51mmol)加入搅拌的2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃(结构式3所示化合物)(50mmol)和4
‑
甲氧基苯甲酰氯(110mmol)的二氯甲烷(50ml)溶液中。将混合物在相同温度下搅拌0.5小时,恢复室温后继续搅拌2小时。温度再降至0
‑
5℃,将水(50ml)逐滴加入搅拌的混合物中。然后用二氯甲烷(3
×
30ml)萃取混合物。将合并的有机层用饱和碳酸氢钠水溶液洗涤,用无水硫酸钠干燥并浓缩。通过柱色谱法(洗脱剂:乙酸乙酯/己烷1:3体积比)纯化后,得到16.91g淡黄色固体,产率62.8%。
[0043]1h
‑
nmr(400mhz,cdcl3)δ:0.90(t,6h),1.30(m,4h),1.59(m,4h),3.18(t,4h),3.79(s,6h),7.10(d,4h),7.54(s,1h),7.73
‑
7.97(m,5h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,22.40,27.95,29.26,56.08,96.39,114.40,117.53,118.58,118.69,129.77,134.06,159.33,164.47,172.69,193.50.lc
‑
ms(esi,pos,ion)m/z:539.25[m+h].
[0044]
(4)中间体(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮)(结构式5所示化合物)的合成
[0045]
将中间体(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
甲氧基苯基)甲酮)(结构式4所示化合物)(50mmol)加入到hbr(10mmol)的acoh(100ml)溶液中,回流反应15小时。将反应混合物冷却并缓慢倒入冷水(80ml)中,保持温度低于15℃并用dcm(100ml)萃取。将得到的dcm层用水(80ml)洗涤,然后用5%的nahco3(100ml)溶液洗涤。无水硫酸镁干燥有机相,浓缩至干,得到粗品,用正庚烷(60ml)打浆,过滤,干燥,得到22.77g浅棕色固体,产率89.2%。
[0046]1h
‑
nmr(400mhz,cdcl3)δ:0.90(t,6h),1.30(m,4h),1.59(m,4h),2.91(t,4h),5.00(s,2h),6.88(d,4h),7.68(d,4h),7.78(s,1h),8.04(s,1h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,22.40,27.95,29.26,96.39,115.77,117.53,118.58,118.69,129.41,133.29,159.33,163.59,172.69,193.50.lc
‑
ms(esi,pos,ion)m/z:511.22[m+h].
[0047]
(5)(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
(3
‑
(二丁基氨基)丙氧基)苯基)甲酮)(结构式6所示化合物)的合成
[0048]
将碳酸钾(20mmol)加入到(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮)(结构式5所示化合物)(20mmol)的甲苯(80ml)溶液中。然后将混合物在25
‑
30℃下搅拌0.5小时并加热至回流,随后保持温度加入1
‑
氯
‑3‑
二正丁基氨基丙烷(45mmol)。将反应混合物回流反应5小时。然后将反应混合物冷却至25
‑
30℃,过滤,用甲苯(50ml)洗涤。收集滤液并减压浓缩,快速柱色谱分离(洗脱剂用乙酸乙酯),得到15.20g类白色固体,产率89.5%。
[0049]1h
‑
nmr(400mhz,cdcl3)δ:0.87
‑
0.94(m,18h),1.21
‑
1.34(m,12h),1.35
‑
1.48(m,8h),1.59(m,4h),1.83(m,4h),2.38(t,8h),2.48(t,4h),2.91(t,4h),4.05(t,4h),7.12(d,4h),7.76(s,2h),7.84(d,4h).
13
c
‑
nmr(125mhz,cdcl3)δ:14.00,20.75,22.40,27.95,28.13,29.25,29.26,51.76,56.45,67.16,96.39,115.25,117.53,118.58,118.69,129.90,134.17,159.33,165.76,172.69,193.50.lc
‑
ms(esi,pos,ion)m/z:849.55[m+h].
[0050]
实施例2、化合物2(化学结构式7所示化合物)的制备
[0051]
(1)(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
((2
‑
甲基丙烯基)氧基)苯基)甲酮)(化学结构式7所示化合物)的合成:
[0052][0053]
将碳酸钾(20mmol)加入到(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮)(结构式5所示化合物)(20mmol)的甲苯(80ml)溶液中。然后将混合物在25
‑
30℃下搅拌0.5小时并加热至回流,随后保持温度加入3
‑
氯
‑3‑
甲基丙烯(45mmol)。将反应混合物回流反应5小时。然后将反应混合物冷却至25
‑
30℃,过滤无机固体并用甲苯(50ml)洗涤。收集滤液并减压浓缩,快速柱色谱分离(洗脱剂用乙酸乙酯),得到9.71g类白色固体,产率为78.5%。
[0054]1h
‑
nmr(400mhz,cdcl3)δ:0.91(t,6h),1.28(m,4h),1.67(m,4h),1.87(s,6h),2.75(t,4h),4.39(s,4h),5.00(s,4h),7.02(m,4h),7.44(s,1h),7.82(m,5h).
13
c
‑
nmr(125mhz,cdcl3)δ:13.89,19.16,20.84,26.99,27.68,71.38,93.09,112.42,114.86,117.6,123.65,130.12,132.81,139.43,155.21,162.37,162.76,192.09.lc
‑
ms(esi,pos,ion)m/z:619.30[m+h].
[0055]
实施例3、化合物3(结构式8所示化合物)的制备
[0056]
(1)(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
(2
‑
甲基丁氧基)苯基)甲酮)(结构式8所示化合物)的合成
[0057][0058]
将碳酸钾(20mmol)加入到(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮)(结构式5所示化合物)(20mmol)的甲苯(80ml)溶液中。然后将混合物在25
‑
30℃下搅拌0.5小时并加热至回流,随后保持温度加入1
‑
氯
‑2‑
甲基丁烷(45mmol)。将反应混合物回流反应5小时。然后将反应混合物冷却至25
‑
30℃,过滤无机固体并用甲苯(50ml)洗涤。收集滤液并减压浓缩,快速柱色谱分离(洗脱剂用乙酸乙酯),得到10.74g类白色固体,产率为82.5%。
[0059]1h
‑
nmr(400mhz,cdcl3)δ:0.90(m,12h),1.05(d,6h),1.27(m,4h),1.51(m,4h),
1.67(m,4h),1.97(m,2h),2.75(t,4h),3.94(m,4h),6.98(dt,4h),7.44(s,1h),7.83(m,5h).
13
c
‑
nmr(125mhz,cdcl3)δ:11.48,13.81,17.5,22.28,26.75,28,31.86,35.26,72.75,96.81,117.86,120.03,122.43,129.79,136.79,142.43,154.73,158.16,192.49.lc
‑
ms(esi,pos,ion)m/z:651.37[m+h].
[0060]
实施例4、化合物4(结构式9所示化合物)的制备
[0061]
(1)(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
(环戊基氧基)苯基)甲酮)(结构式9所示化合物)的合成
[0062][0063]
将碳酸钾(20mmol)加入到(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮)(结构式5所示化合物)(20mmol)的甲苯(80ml)溶液中。然后将混合物在25
‑
30℃下搅拌0.5小时并加热至回流,随后保持温度加入环戊基氯(45mmol)。将反应混合物回流反应5小时。然后将反应混合物冷却至25
‑
30℃,过滤无机固体并用甲苯(50ml)洗涤。收集滤液并减压浓缩,快速柱色谱分离(洗脱剂用乙酸乙酯),得到11.36g类白色固体,产率为87.8%。lc
‑
ms(esi,pos,ion)m/z:647.32[m+h].
[0064]
实施例5、化合物5(结构式10所示化合物)的制备
[0065]
(1)(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
(苯基乙氧基)苯基)甲酮)(结构
[0066]
式10所示化合物)的合成
[0067][0068]
将碳酸钾(20mmol)加入到(2,6
‑
二丁基苯并[1,2
‑
b:5,4
‑
b']二呋喃
‑
3,5
‑
二基)双((4
‑
羟基苯基)甲酮)(结构式5所示化合物)(20mmol)的甲苯(80ml)溶液中。然后将混合物在25
‑
30℃下搅拌0.5小时并加热至回流,随后保持温度加入1
‑
氯
‑2‑
苯基乙烷(45mmol)。将反应混合物回流反应5小时。然后将反应混合物冷却至25
‑
30℃,过滤无机固体并用甲苯(50ml)洗涤。收集滤液并减压浓缩,快速柱色谱分离(洗脱剂用乙酸乙酯),得到11.44g类白色固体,产率为79.6%。lc
‑
ms(esi,pos,ion)m/z:719.33[m+h].
[0069]
生物学实施例1、本发明所得化合物对akt
‑
1激酶活性的影响
[0070]
本发明所得化合物对akt激酶的活性影响采用下述方法测定:该方法描述了一种激酶测定,其采用市售imap试剂盒(imap akt assay bulk kit,#r8059,molecular devices,sunnyvale,ca),通过荧光极化测量全长人重组活性akt
‑
1对荧光标记肤的磷酸化。所用的akt
‑
1激酶由用pdk1和map激酶2活化的全长人重组akt
‑
1制备。测定程序始于dmso中10mm化合物贮液的制备。2倍梯度稀释9次,按照试剂盒的操作,将不同浓度梯度的化合物和阳性对照(akti
‑
1/2)混合相应缓冲底物,加入200nm荧光标记肽底物和4nm makt
‑
1置于384孔板上启动测定。将板以100g离心1分钟后,置于室温下保持60分钟。然后加入结合溶液淬灭反应,再次离心,于室温下再保持30分钟,然后在victor 1420mutilabel hts计数器上读数,以测量荧光极化。结果如图1所示,本发明所得化合物显示出优秀的akt蛋白激酶抑制活性。
[0071]
生物学实施例2、本发明所得化合物对肿瘤细胞增殖的影响
[0072]
本发明所得化合物的抗肿瘤细胞增殖活性使用mtt法测定。卵巢癌skov3和人肝癌smmc
‑
7721细胞培养在含有10%胎牛血清和1%青霉素/链霉素的rpmi1640培养基中培养。细胞培养在37℃,5%co2的细胞培养箱中。培养好的细胞用胰蛋白酶消化后,转移至96孔板上(2000细胞/孔)。继续培养过夜后,将本发明所得化合物以指定的工作浓度处理细胞。孵育48小时后,将20ul mtt(5mg/ml)滴入孔中。继续孵育4小时后,去除96孔板中的混合培养基,然后添加150ul二甲基亚砜(dmso)。接下来,将96孔板摇床约15分钟,保持室温以混合其内容物。然后在490nm波长下测量od值。结果见图2。结果表明本发明所得化合物对卵巢癌细胞和肝癌细胞的增殖具有抑制作用。
[0073]
生物学实施例3、蛋白质免疫印迹法对akt激酶信号通路的影响。
[0074]
培养好的skov3细胞用胰蛋白酶消化后,转移至6孔板上(1x106细胞/孔)。继续培养过夜后,将本发明所得化合物以100um的工作浓度处理细胞。处理24小时,收获细胞。培养的细胞收获后,使用全细胞裂解液(whole cell lysates,wcl)裂解后离心,加入样品缓冲液煮沸,进行sds
‑
page电泳,电泳结束后将蛋白转移到pvdf膜上,5%bsa封闭后,一抗akt、p
‑
akt(ser473)和p
‑
akt(thr308)和内参蛋白(gapdh))孵育过夜,冲洗后hpr
‑
标记的二抗室温孵育两个小时,冲洗后化学发光法检测enhanced chemiluminescence(ecl)detection system。结果如附图3,本发明所制备的化合物通过可能通过抑制pi3k/akt信号通路的活化,发挥诱导细胞凋亡的作用。
[0075]
以上说明本发明所制备的化合物能够有效抑制akt蛋白激酶活性,可以作为一种akt蛋白激酶抑制剂来有效地预防和/或治疗过度增生性疾病如癌症的方法。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1