一种基于单核苷酸多态性的肺血栓栓塞症风险预测模型的构建方法、SNP位点组合及应用与流程
一种基于单核苷酸多态性的肺血栓栓塞症风险预测模型的构建方法、snp位点组合及应用
技术领域
1.本发明涉及一种基于单核苷酸多态性的肺血栓栓塞症风险预测模型的构建方法、snp位点组合及应用,具体涉及肺血栓栓塞症相关的单核苷酸多态性位点及其在风险预测中的用途,属于疾病检测技术领域。
背景技术:
2.肺血栓栓塞症(pulmonary thromboembolism,pte)是一种由于产生静脉血栓,堵塞肺动脉或其分支,引起肺循环障碍的疾病,具有潜在的致死风险。pte在住院病人中常见,且发病率及死亡率较高。导致pte发生的危险因素主要包括感染、骨折/创伤、吸烟、恶性肿瘤、肥胖、妊娠等。pte起病隐匿,大多数的pte患者的临床表现(如胸闷、憋气、咳嗽、气促、下肢水肿等)缺乏特异性,常常由于原发病的缘故而忽略掉这些非特异性的表现,故在一些慢性病程者容易造成漏诊误诊,进而造成严重后果。临床研究显示,合理预防措施可使dvt相对风险降低50%
‑
60%,pte相对风险降低近2/3。有效的风险预测对预防pte的发生、降低患者死亡率至关重要,改善风险评估和预测工具是减少pte疾病发生的重要措施。因此,急需一种可靠、便捷的方法进行风险预测,以筛选可能发生pte的高危患者,从而进行必要的预防干预措施,比如抗凝,以便最大程度降低pte的发生率和死亡率。
3.目前,临床上pte的预测暂缺乏较好的血清学标志物,尽管已有研究证实:d二聚体、c反应蛋白、组织因子、凝血因子viii及血小板、白细胞、血小板等可以在一定程度上反映患者的高凝状态,提示患者pte的发生,但其预测价值有限,实际临床工作中应用并不多。并且,我国对这类疾病认识及研究起步晚,在pte风险评估模型方面上也大多是借用引进国外研制的,如caprini血栓评估模型、padua预测评分模型、autar血栓评估模型和kucher量表。但是由于存在人种、体质、文化及生活习惯等方面的差异,在使用上也有一定的局限性,如凝血因子v leiden突变、凝血酶原g20210a突变在高加索人种中的发生率较高,但几乎不存在于中国人群中。而亚洲人群中蛋白c、蛋白s的缺失或抗凝血酶的缺乏发生率高于高加索人种,但并未在caprini量表中有所体现。并且上述量表大多条目复杂,如caprini包含有将近40项评估条目,实施过程耗时耗力,不便于临床工作者使用。
4.另外,肺血栓栓塞症的发生是遗传易感性和获得性危险因素相互作用的结果。家族性研究发现pte的遗传度大约为50%
‑
60%。也就是说,个体在遭受vet相关危险因素后,后续临床发展将很大程度上取决于个体的遗传易感性。单核苷酸多态性(single nucleotide polymorphisms,snp)是指在基因组上单个核苷酸的变异,包括转换、颠换、缺失和插入而形成的遗传标记,其数量很多,多态性丰富,与很多人体表型、对药物或疾病的易感性等相关。因此,snp是导致个体疾病发生发展差异的重要遗传基础。并且,一些基因多态性位点在不同种族中的分布不同,而之前的大部分相关研究是在高加索人群中完成的,因为两个种族的基因频率并不完全相同,其研究结果可能不适用于汉族患者。
5.综上,一个理想的pte风险评估模型是经得起其他的临床实验进行验证的,可以准
确识别出pte的高危人群,从而提高血栓预防率和降低pte的发生率。但它不会包含太多的条目,以便可广泛应用于在日常的临床实践中。目前亟需开发一种适于亚种人群,特别是中国人群的snp位点及位点组合模式用于vet的早期风险筛查,且基于这些snp位点,可以实现对个体pte患病风险的预测。
技术实现要素:
6.针对上述现存的技术问题,本发明提供一种基于单核苷酸多态性的肺血栓栓塞症风险预测模型的构建方法、snp位点组合及应用,开发有效科学、国人适用的pte风险评估模型,将更加有效地降低pte发生率和病死率,减少后遗症,提高pte患者的生活质量,从而全面提升医疗质量与安全,减少卫生资源的消耗。
7.为实现上述目的,首先,本发明提供一种基于单核苷酸多态性的肺血栓栓塞症发生风险预测模型的构建方法,包括如下具体步骤:
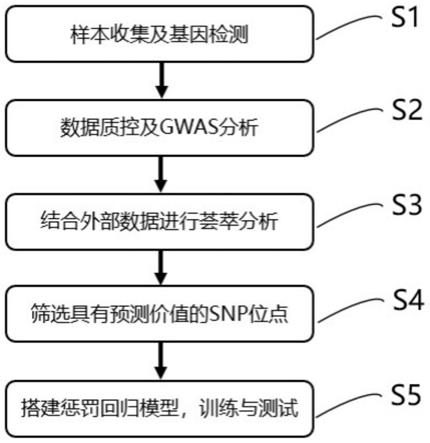
8.s1、样本收集及基因检测:
9.(1
‑
1)招募肺血栓栓塞症患者,以及作为对照的健康个体。
10.上述技术方案中,要求两组招募对象的年龄,性别和血统等结构信息相匹配,以排除混杂因素。并且招募对象均来自于中国汉族人群,使得分析得到的snp位点可以准确的反应中国人群的pte患病风险。
11.(1
‑
2)收集招募对象的外周血液样本,使用高通量基因芯片技术进行基因检测,得到所有招募对象的基因组数据。
12.gwas常用的高通量基因分型手段有基因芯片技术、全基因组重测序和全基因组外显子测序等。全基因组重测序对基因组遗传信息挖掘全面,但数据量大,成本较高。全基因组外显子测序极大降低了待测序列总量,但并未过多降低遗传信息。基因芯片可以实现对特定群体特定snp位点的快速分型。较低的分型成本、时间成本、储存成本和分析成本是目前基因芯片技术的优势所在。上述技术方案中,基因检测使用的illumina wegene v2芯片是专门针对中国人群遗传背景设计,全面覆盖中国人群体中发现的常见变异,包含了策略性选择并优化的700,000多个标签snp。
13.s2、数据质量控制及全基因组关联分析(gwas):
14.(2
‑
1)对步骤s1得到的基因组数据进行样本的质量控制,得到合格样本的基因组数据。
15.进一步,所述的样本质量控制包括:删除性别错误的个体,因为通过x染色体杂合率计算性别,性别错误的个体可能存在dna污染的问题;
16.删除snp缺失率过高的个体,阈值为0.05。因为snp缺失率过高说明dna质量及分型质量不好;
17.删除有亲缘关系的样本,亲缘关系可以用identity by state(ibs)来衡量,设置阈值为0.2。
18.(2
‑
2)对步骤s1得到的合格样本的基因组数据进行位点的质量控制,得到合格的snp位点。
19.进一步,所述的位点的质量控制:删除缺失率过高的snp位点,剔除缺失率在20%以上的位点;
20.删除等位基因频率(maf)较小的snp位点,阈值为0.01;
21.删除偏离哈迪温伯格平衡的snp位点(hwe)(p<1
×
10
‑5)。
22.(2
‑
3)对步骤s2
‑
2得到的合格的snp位点进行基因型填充(imputation),首先采用eagle软件构建单倍型,然后采用minimac4软件进行填补,以1000genomes计划phase3的基因型数据作为参照,再针对填补后的位点进行质量控制。
23.进一步,所述的对填补后的位点进行质量控制包括:
24.删除填充质量(imputation quality)较小的位点,阈值为0.3;
25.删除等位基因频率(maf)较小的snp位点,阈值为0.01;
26.删除缺失率过高的snp位点,剔除缺失率在20%以上的位点。
27.(2
‑
4)基于合格样本的基因组数据,随机选取其中的80%作为gwas分析和模型训练的数据,另外20%单纯作为测试数据,以将特征选择过程与模型测试过程相互独立。
28.上述技术方案将特征选择过程与测试过程相互独立,能够避免模型过拟合,提高泛化能力。
29.(2
‑
5)基于选取的用于gwas分析的基因组数据,采用逻辑回归模型,并对年龄,性别和群体结构分析中的前五个主成分进行协变量校正,通过gwas分析来检测与pte显著关联的位点,以p值来衡量关联标记的显著性。
30.进一步,所述的群体结构的主成分分析使用软件为gcta v.1.91,关联分析采用plink1.9软件,p<5
×
10
‑8视为具有统计学意义。
31.上述技术方案中,gwas研究的基本原理是:在一定人群中选出病例组与对照组,比较所有snp位点的等位基因频率在两组之间的差异,如果某个位点的等位基因频率在对照组中明显高于或低于病例组,则该位点极有可能与疾病存在关联,然后即可根据其在基因组中的位置和连锁不平衡关系推测出可能的致病基因。
32.为了避免因群体分层现象而导致出现假阳性或假阴性结果,基于位于常染色体上的变异位点,运用gcta v.1.91软件基于主成分分析估计人群分层情况,并将最显著的主成分特征向量作为协变量纳入模型。分析样本的群体结构可以有效的降低标记与目标性状的伪关联程度,提高关联分析的准确性。
33.s3、结合外部人群的基因组数据进行荟萃分析(meta analysis):
34.(3
‑
1)从数据库中获取外部人群基因组数据的概括统计(summary statistic)数据,并对外部人群的基因组数据进行gwas分析,方法如步骤s2中所述,得到显著相关的snp位点。
35.(3
‑
2)针对外部人群基因组数据和所有招募对象的基因组数据,采用混合效应模型方法进行荟萃分析,根据p值选取显著关联的snp位点,以提高检验效能,有效检测相关联的snp位点。
36.上述技术方案中,荟萃分析指将多个独立的研究数据集组合在一起最终提供一个总计结果的一种统计学分析方法,目的是通过增大样本含量来提高检验效能,增加结论的可信度,有效检测相关联的低频和罕见变异位点。
37.s4、筛选具有预测价值的snp位点组合:
38.针对步骤s3
‑
2荟萃分析得到的snp位点,采用异质性检验(cochran's q test),剔除在外部人群和招募对象中异质性较高的snp位点,得到具有预测价值的snp位点组合。该
snp位点组合能够预测pte,可以作为临床检测的panel。
39.s5、搭建回归模型,进行训练与测试:
40.(5
‑
1)使用r语言bigstatsr包,建立惩罚线性回归模型,选择最佳超参数。
41.进一步,本发明最佳超参数的选择使用网格搜索结合交叉验证的方法,通过网格搜索使用每组超参数训练模型,挑选出验证集误差最小的超参数,作为最好的超参数。
42.上述技术方案中,超参数会影响算法运行的时间和存储成本,有些超参数会影响学习到的模型质量以及在新输入上推断正确结果的能力,因此超参数的选择至关重要。
43.(5
‑
2)基于步骤s2中选取的训练数据,对模型进行训练,得到各特征相应的回归系数。
44.(5
‑
3)基于步骤s2中选取的测试数据,对模型进行性能测试,绘制roc曲线,计算曲线下面积。
45.其次,本发明又提供一种根据上述肺血栓栓塞症风险预测模型的构建方法得到的具有预测价值的snp位点组合,包括如下表中的至少一种:
46.47.[0048][0049]
其中,rsid表示snp位点编号;chr代表所在染色质区;pos代表位点所在位置,基于hg19/grch37参考基因组;ref表示参考基因组的基因型;alt代表风险等位基因。
[0050]
上述技术方案中,基于中国人群的gwas研究,筛选得到pte相关的单核苷酸多态性位点组合,更适合亚洲人群,特别是在中国人群中进行肺血栓栓塞症的早期风险筛查。
[0051]
再者,本发明再提供一种上述snp位点组合作为肺血栓栓塞症风险评估方面的应用或筛查产品中的应用。
[0052]
综上,本发明提供了肺血栓栓塞症相关联的单核苷酸多态性位点及基于snp位点开发的肺血栓栓塞症风险预测模型,具体而言是以筛选的snp位点为基础,采用惩罚回归模型,实现对个体发生肺血栓栓塞症的风险预测。本发明方法构建的肺血栓栓塞症风险预测模型,能够预测pte患病风险,相比于血清标志物或其它评估模型具有更高的准确率,snp位点组合更适合亚洲人群,特别是在中国人群中进行肺血栓栓塞症的早期风险筛查,可以辅助医务人员识别出临床上存在的高危患者,进行必要的预防与干预措施,降低肺血栓栓塞症的发生率和死亡率,提升医疗质量。
[0053]
相比现有技术,本发明具有如下技术优势:
[0054]
1、样本数据方面:一些基因多态性位点在不同种族中的分布不同,之前的研究均是在高加索人群中完成的,因为两个种族的基因频率不完全相同,因此其分析得到的snp位点并不等同于是中国人群的风险位点,这些位点可能与中国人群患pte并无相关性。然而,本发明研究收集到的病例均来自于中国汉族人群,分析得到的snp位点准确的反应了中国人群的患病风险,并基于中国人群进行训练与测试,使得模型更加适合中国人群使用,测试结果更加贴近真实情况。
[0055]
2、基因芯片方面:本发明使用的illumina wegene v2芯片,是专门针对中国人群
遗传背景设计,使用超过十万中国人群数据作为芯片设计的参考数据,全面覆盖中国人群体中发现的常见变异,包含了策略性选择并优化的700,000多个标签snp。芯片骨架采用全新骨架设计优化算法,显著提升中国人群imputation效果。临床相关变异采用来自最新数据库的变异位点(包括clinvar,acmg 59,cpic,pharmgkb和nhgri
‑
ebi gwas catalog),适用于临床疾病风险的预测。
[0056]
3、gwas分析方面:本发明结合了外部人群的基因组数据进行荟萃分析,包含4620例pte病例和356,574例健康对照。将外部数据与发明人收集的中国人群数据分别独立gwas分析之后,再采用混合效应模型方法进行荟萃分析,目的是通过增大样本含量来提高检验效能,增加结论的可信度,可以有效检测相关联的低频和罕见变异位点。
[0057]
4、模型构建方面:在构建线性回归模型时,本发明采用网格搜索的方法来寻找最佳惩罚项,以提高模型性能。另外,本发明基于训练集进行特征选择,测试集仅用于测试,将特征选择过程与测试过程相互独立,可以避免模型过拟合问题,使测试结果更加符合实际使用情况。
附图说明
[0058]
图1为本发明肺血栓栓塞症风险预测模型的构建方法的步骤流程图;
[0059]
图2为本发明实施例中测试模型得到的roc曲线。
具体实施方式
[0060]
下面结合附图和实施例对本发明作进一步说明,而不是对本发明的限制。
[0061]
实施例1:本发明肺血栓栓塞症风险预测模型的构建方法,如图1所示,包括如下具体步骤:
[0062]
s1、样本收集及基因检测:
[0063]
(1
‑
1)从中国肺栓塞登记研究(china pulmonary thromboembolism registry study,cures)中招募到1237例肺栓塞病例(cases),并获得所有研究参与者的知情同意和中日友好医院伦理委员会的批准。参照病例组的年龄,性别和血统等结构信息,从微基因公司数据库中随机选择3873例健康个体作为对照(controls)。具体实施时,要求两组招募对象的年龄,性别和血统等结构信息相匹配,且均来自于中国汉族人群。
[0064]
(1
‑
2)采集招募者的外周血液样本,提取dna,将质检合格的dna样本使用illumina高通量基因芯片技术进行基因分型检测,获得cases和controls的基因组数据。具体实施时,基因检测使用illumina wegene v2芯片,但本发明不受限于使用其它基因检测方法进行检测。
[0065]
s2、数据质量控制及全基因组关联分析(gwas):
[0066]
(2
‑
1)样本的质量控制:针对cases和controls的基因组数据,删除性别错误的个体;删除snp缺失率过高的个体,设置阈值为0.05;删除有亲缘关系的样本,亲缘关系可以用identity by state(ibs)来衡量,设置阈值为0.2。
[0067]
经过样本的质量控制后,得到1181例pte病例和3749例健康对照的合格样本的基因组数据。
[0068]
(2
‑
2)位点的质量控制:针对步骤s2
‑
1得到的合格样本的基因组数据,删除缺失率
过高的snp位点,剔除缺失率在20%以上的位点;删除等位基因频率(maf)较小的snp位点,设置阈值为0.01;删除偏离哈迪温伯格平衡的snp位点(hwe)(p<1
×
10
‑5)。
[0069]
(2
‑
3)基因型填充:经过步骤s2
‑
2对snp位点初步的质量控制后,接着进行基因型填充(imputation)。首先采用eagle软件构建单倍型,然后采用minimac4软件进行填补,以1000genomes计划phase3的基因型数据作为参照,再针对填补后的snp位点进行进一步的质量控制:删除填充质量(imputation quality)较小的位点,阈值为0.3;删除等位基因频率(maf)较小的snp位点,阈值为0.01;删除缺失率过高的snp位点,剔除缺失率在20%以上的位点。
[0070]
经过位点的质量控制后,共得到大约七百万个变异位点,可以用于后续的gwas分析。
[0071]
(2
‑
4)数据选取;针对合格样本的基因组数据,随机选取其中的80%作为gwas分析和模型训练的数据,另外20%单纯作为测试数据,从而将特征选择过程与模型测试过程相互独立。如此共有3945个合格样本的基因组数据可以作为训练集,以进行gwas分析。
[0072]
(2
‑
5)gwas分析:为了避免因群体分层现象而导致出现假阳性或假阴性结果,基于位于常染色体上的变异位点,gwas采用逻辑回归模型,并对年龄,性别和群体结构分析中的前五个主成分进行协变量校正,通过全基因组关联分析来检测与pte显著关联的位点,以p值来衡量关联标记的显著性。具体实施时,群体结构的主成分分析使用软件为gcta v.1.91,关联分析采用plink 1.9软件,p<5
×
10
‑8视为具有统计学意义。
[0073]
s3、结合外部人群的基因组数据进行荟萃分析(meta analysis):
[0074]
(3
‑
1)从英国生物样本库(uk biobank)等数据库中获取外部人群基因组数据的概括统计(summary statistic)数据,包含4620例pte病例和356,574例健康对照。对外部人群的基因组数据进行gwas分析,方法如步骤s2中所述,获得显著关联的snp位点。
[0075]
(3
‑
2)将外部人群基因组数据与步骤s1收集的中国人群的基因组数据分别独立进行gwas分析之后,再采用混合效应模型方法进行荟萃分析,并根据p值选取显著关联的snp位点。目的是通过增大样本含量来提高检验效能,有效检测相关联的变异位点。
[0076]
s4、筛选具有预测价值的snp位点组合:
[0077]
(4
‑
1)针对步骤s3
‑
2荟萃分析选取的具有显著性的snp位点,使用异质性检验(cochran's q test),剔除在外部人群和招募对象中异质性较高的位点。
[0078]
(4
‑
2)经过异质性检验后,得到48个显著关联的snp位点组合,该snp位点组合具有pte预测价值,可以作为临床检测的panel。
[0079]
具体的,上述48个vet易感snp位点组合的信息如下表1:
[0080]
表1
[0081]
[0082]
[0083][0084]
注:rsid表示snp位点编号;chr代表所在染色质区;pos代表位点所在位置(基于hg19/grch37参考基因组);ref表示参考基因组的基因型;alt代表风险等位基因;prs beta表示该snp位点在本实施例肺血栓栓塞症风险预测模型中的回归系数。
[0085]
并且,包括上表中至少一种的snp位点组合,均可以作为肺血栓栓塞症风险评估方面的应用或筛查产品中的应用。
[0086]
s5、搭建回归模型,进行训练与测试:
[0087]
(5
‑
1)使用r语言bigstatsr包,建立惩罚线性回归模型,使用网格搜索结合交叉验证的方法,通过网格搜索使用每组超参数训练模型,挑选出验证集误差最小的超参数,作为最好的超参数。
[0088]
(5
‑
2)基于步骤s2中随机选取的训练数据,对模型进行训练,得到各特征相应的回归系数。
[0089]
(5
‑
3)基于步骤s2中随机选取的测试数据,对模型进行性能测试,绘制roc曲线,得到曲线下面积auc=0.666,如图2所示。
[0090]
由上可知,本发明提供建模方法可以搭建肺血栓栓塞症风险预测模型,获得snp位点组合,从而预测pte患病风险,预判是否会产生肺血栓栓塞症,进而个性化有针对性的用药,实现精准医疗。
[0091]
实施例2:关于本发明肺血栓栓塞症风险预测模型的构建方法和snp位点组合的应用,例如要对某位临床患者进行pte风险预测,将按照如下步骤进行实施:
[0092]
(a)采集该患者的外周静脉血液,对血液进行抗凝操作。
[0093]
(b)从采集的外周血中提取基因组dna,并进行质检和浓度测定。
[0094]
(c)质检合格后,进行遗传易感位点检测,检测方法可以使用基因芯片或二代测序,检测内容为表1所示的48个vet易感snp位点组合,此步骤可以实现对易感位点进行基因
分型。
[0095]
(d)根据实施例1构建的回归模型,各项特征的回归系数,利用基因分型结果,计算风险分数,根据风险分数可以得知该患者的pte易感风险,继而根据患病风险及时采取相应的预防措施。
[0096]
由上可知,相比较于其他单分子标记物,本方法获得的肺血栓栓塞症风险预测模型具有更高的准确率和精度,相比于其它预测模型更适合亚洲人群,特别是中国人群,并且检测评估方法更简洁高效,更便于临床实践,从而降低肺血栓栓塞症的发生率和死亡率,提升医疗质量。
[0097]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1