一种抗HIV类药物中间体晶体的生长方法和所得晶体及其应用与流程
一种抗hiv类药物中间体晶体的生长方法和所得晶体及其应用
技术领域
1.本发明属于生物医药技术领域,具体涉及一种抗hiv类药物中间体晶体的生长方法和所得晶体及其应用。
背景技术:
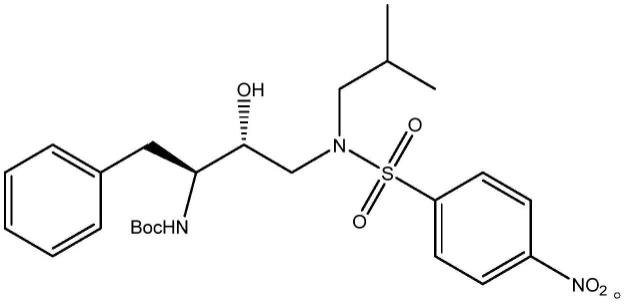
2.叔丁基(2s,3r)-3-羟基-4-(n-异丁基-4-硝基苯磺胺基)-1-苯丁烷-2-氨基甲酸酯可用于制备福沙那韦钙、地瑞那韦、安瑞那韦等抗hiv类药物,是一种重要中间体。其结果如下所示:
[0003][0004]
其中,福沙那韦钙的合成路线如下所示:
[0005][0006]
其中,安瑞那韦的合成路线如下所示:
[0007][0008]
其中,地瑞那韦的合成路线如下所示:
[0009][0010]
作为一种可以制备多种api的关键中间体,叔丁基(2s,3r)-3-羟基-4-(n-异丁基-4-硝基苯磺胺基)-1-苯丁烷-2-氨基甲酸酯的晶型的稳定性有利于减少在进行下一步反应时因在贮存过程中产生的杂质,提高合成的产率和质量。
[0011]
专利wo2006131757公开了叔丁基(2s,3r)-3-羟基-4-(n-异丁基-4-硝基苯磺胺基)-1-苯丁烷-2-氨基甲酸酯在进一步反应前需要分离、转移、储存,但其产物晶型外观为纤维状,难以过滤,容易留有溶剂,造成转移困难,,引起其他处理困难。
[0012]
专利wo2006131757公开的叔丁基(2s,3r)-3-羟基-4-(n-异丁基-4-硝基苯磺胺基)-1-苯丁烷-2-氨基甲酸酯的制备方法是通过加热饱和溶液后冷却得到晶体,或者在冷却过程中引晶得到晶体。最后得到的晶型的稳定性较差。
技术实现要素:
[0013]
针对现有技术的不足,本发明的目的在于提供一种抗hiv类药物中间体晶体的生长方法和所得晶体及其应用。
[0014]
为达到此发明目的,本发明采用以下技术方案:
[0015]
第一方面,本发明提供一种抗hiv类药物中间体晶体的生长方法,其特征在于,所述生长方法包括如下步骤:
[0016]
将原料在55-65℃下溶解于甲醇中,过滤,然后将滤液以0.05-0.15℃/min的速度
冷却至20-30℃,搅拌2-3h,分离得到晶体;
[0017]
所述原料为抗hiv类药物中间体粗品;
[0018]
所述抗hiv类药物中间体的结构如式ⅰ所示:
[0019][0020]
本发明所涉及的抗hiv类药物中间体晶体的生长方法简单易操作,且制得的式ⅰ所示化合物晶体外观呈棒状型固体,有助于其纯化操作;晶体稳定,有利于减少在进行下一步反应时因在贮存过程中产生的杂质,提高合成的产率和质量;在进一步制备抗hiv类药物的反应中,该晶体还能够显著提高反应速率。
[0021]
上述55-65℃例如可以是55℃、56℃、57℃、58℃、59℃、60℃、61℃、62℃、63℃、64℃或65℃等,该范围内的其他具体点值均可选择,在此便不再一一赘述。
[0022]
上述0.05-0.15℃/min例如可以是0.05℃/min、0.06℃/min、0.07℃/min、0.08℃/min、0.09℃/min、0.10℃/min、0.11℃/min、0.12℃/min、0.13℃/min、0.14℃/min或0.15℃/min等,该范围内的其他具体点值均可选择,在此便不再一一赘述。
[0023]
所述降温速度特定选择为0.05-0.15℃/min是因为若速度进一步提高会出现纤维形态的结晶。
[0024]
上述20-30℃例如可以是20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃或30℃等,该范围内的其他具体点值均可选择,在此便不再一一赘述。
[0025]
优选地,所述原料与甲醇的质量体积比为:每500mg原料溶解于4-12ml(例如4ml、5ml、5.5ml、6ml、6.5ml、7ml、8ml、9ml、10ml、11ml、12ml等,该范围内的其他具体点值均可选择,在此便不再一一赘述)甲醇中。优选地,所述原料与甲醇的质量体积比为:每500mg原料溶解于4-8ml甲醇中。优选地,所述原料与甲醇的质量体积比为:每500mg原料溶解于5.5-6.5ml甲醇中。
[0026]
优选地,所述冷却的速度为0.09-0.11℃/min。
[0027]
优选地,所述滤液冷却至24-26℃。
[0028]
优选地,所述将原料溶解于甲醇中在58-62℃下进行。
[0029]
第二方面,本发明提供一种如上所述的抗hiv类药物中间体晶体的生长方法制得的式ⅰ所示化合物晶体a,所述式ⅰ所示化合物晶体a的x射线衍射图中在8.38
±
0.2
°
、9.44
±
0.2
°
、13.80
±
0.2
°
、16.56
±
0.2
°
、18.82
±
0.2
°
、19.08
±
0.2
°
、19.62
±
0.2
°
、23.58
±
0.2
°
处有特征衍射峰。
[0030]
该晶体外观呈棒状型固体,有助于其纯化操作;晶体稳定,有利于减少在进行下一步反应时因在贮存过程中产生的杂质,提高合成的产率和质量。
[0031]
第三方面,本发明提供另一种抗hiv类药物中间体晶体的制备方法,所述制备方法为:将如上所述的式ⅰ所示化合物晶体a加热至100-170℃(例如100℃、110℃、115℃、120℃、
125℃、130℃、140℃、160℃、170℃等),优选加热至115-125℃,即得式ⅰ所示化合物晶体b。
[0032]
所述加热温度特定选择为100-170℃是因为若温度进一步提高或降低都不能制备出特定晶型的目标化合物。
[0033]
第四方面,本发明提供一种如上所述的制备方法制得的式ⅰ所示化合物晶体b,所述式ⅰ所示化合物晶体b的x射线衍射图中在7.09
±
0.2
°
、9.55
±
0.2
°
、10.56
±
0.2
°
、11.48
±
0.2
°
、14.08
±
0.2
°
、16.12
±
0.2
°
、17.61
±
0.2
°
处有特征衍射峰。
[0034]
该晶体外观呈棒状型固体,有助于其纯化操作;晶体稳定,有利于减少在进行下一步反应时因在贮存过程中产生的杂质,提高合成的产率和质量。
[0035]
第五方面,本发明还提供十二种抗hiv类药物中间体晶体的制备方法,所述制备方法为:将如上所述的式ⅰ所示化合物晶体b悬浮于溶剂中,所述溶剂分别为第一溶剂、邻二甲苯、2-丁醇、正丁醇、乙酸、1,2-二氯乙烷、甲酰胺、乙醇、甲酸、1,4-二氧嘧啶、二甲亚砜、吡啶,20-30℃下静置48-96h,分别分离得到式ⅰ所示化合物晶体c、晶体d、晶体e、晶体f、晶体g、晶体h、晶体i、晶体j、晶体k、晶体l、晶体m、晶体n;第一溶剂为正庚烷、甲苯或二氯甲烷中的任意一种。
[0036]
上述48-96h例如可以是48h、55h、60h、65h、70h、75h、80h、85h、90h或96h等,该范围内的其他具体点值均可选择,在此便不再一一赘述。
[0037]
优选地,所述悬浮在20-40℃下进行,例如可以是20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃、30℃、35℃、40℃等,该范围内的其他具体点值均可选择,在此便不再一一赘述。
[0038]
优选地,所述溶解在24-26℃下进行。
[0039]
优选地,所述静置在24-26℃下进行,静置时间为65-75h。
[0040]
优选地,所述晶体b与溶剂的质量体积比为:每50mg晶体b溶解于0.5-1.5ml(例如0.5ml、0.6ml、0.7ml、0.8ml、0.9ml、1.0ml、1.1ml、1.2ml、1.3ml、1.4ml或1.5ml等)溶剂中。
[0041]
优选地,所述晶体b与溶剂的质量体积比为:每500mg晶体b溶解于0.5-1.5ml(例如0.5ml、0.6ml、0.7ml、0.8ml、0.9ml、1.0ml、1.1ml、1.2ml、1.3ml、1.4ml或1.5ml等)溶剂中。
[0042]
由上述制备方法共可获得十二种不同晶型的式ⅰ所示化合物晶体,具体如下所示:
[0043]
第六方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体c,所述式ⅰ所示化合物晶体c的x射线衍射图中在6.96
±
0.2
°
、9.98
±
0.2
°
、10.40
±
0.2
°
、11.04
±
0.2
°
、13.92
±
0.2
°
、16.41
±
0.2
°
、17.97
±
0.2
°
、21.11
±
0.2
°
处有特征衍射峰。
[0044]
第七方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体d,所述式ⅰ所示化合物晶体d的x射线衍射图中在8.68
±
0.2
°
、9.89
±
0.2
°
、15.61
±
0.2
°
、16.07
±
0.2
°
、18.08
±
0.2
°
、20.82
±
0.2
°
、21.69
±
0.2
°
、26.55
±
0.2
°
处有特征衍射峰。
[0045]
第八方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体e,所述式ⅰ所示化合物晶体e的x射线衍射图中在7.30
±
0.2
°
、7.57
±
0.2
°
、8.51
±
0.2
°
、14.56
±
0.2
°
、16.50
±
0.2
°
、18.38
±
0.2
°
、18.75
±
0.2
°
、21.24
±
0.2
°
处有特征衍射峰。
[0046]
第九方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体f,所述式ⅰ所示化合物晶体f的x射线衍射图中在7.32
±
0.2
°
、7.68
±
0.2
°
、8.56
±
0.2
°
、14.64
±
0.2
°
、15.78
±
0.2
°
、16.56
±
0.2
°
、18.94
±
0.2
°
、21.36
±
0.2
°
处有特征衍射峰。
[0047]
第十方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶
体g,所述式ⅰ所示化合物晶体g的x射线衍射图中在7.96
±
0.2
°
、8.68
±
0.2
°
、10.53
±
0.2
°
、14.30
±
0.2
°
、15.26
±
0.2
°
、16.61
±
0.2
°
、17.40
±
0.2
°
、21.70
±
0.2
°
处有特征衍射峰。
[0048]
第十一方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体h,所述式ⅰ所示化合物晶体h的x射线衍射图中在6.38
±
0.2
°
、8.76
±
0.2
°
、9.71
±
0.2
°
、15.93
±
0.2
°
、17.13
±
0.2
°
、18.89
±
0.2
°
、19.96
±
0.2
°
、22.17
±
0.2
°
处有特征衍射峰。
[0049]
第十二方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体i,所述式ⅰ所示化合物晶体i的x射线衍射图中在6.95
±
0.2
°
、8.45
±
0.2
°
、9.56
±
0.2
°
、10.44
±
0.2
°
、13.95
±
0.2
°
、15.20
±
0.2
°
、17.00
±
0.2
°
、17.47
±
0.2
°
处有特征衍射峰。
[0050]
第十三方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体j,所述式ⅰ所示化合物晶体j的x射线衍射图中在7.29
±
0.2
°
、7.55
±
0.2
°
、7.76
±
0.2
°
、8.57
±
0.2
°
、14.64
±
0.2
°
、15.77
±
0.2
°
、16.59
±
0.2
°
、19.01
±
0.2
°
处有特征衍射峰。
[0051]
第十四方面,本发明提供一种如第五方面所述的制备方法如权利要求6所述的制备方法制得的式ⅰ所示化合物晶体k,所述式ⅰ所示化合物晶体k的x射线衍射图中在7.52
±
0.2
°
、8.26
±
0.2
°
、9.36
±
0.2
°
、15.16
±
0.2
°
、16.64
±
0.2
°
、19.23
±
0.2
°
、19.76
±
0.2
°
、23.46
±
0.2
°
处有特征衍射峰。
[0052]
第十五方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体l,所述式ⅰ所示化合物晶体l的x射线衍射图中在6.39
±
0.2
°
、9.53
±
0.2
°
、10.00
±
0.2
°
、15.86
±
0.2
°
、16.51
±
0.2
°
、18.29
±
0.2
°
、19.30
±
0.2
°
、21.24
±
0.2
°
处有特征衍射峰。
[0053]
第十六方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体m,所述式ⅰ所示化合物晶体m的x射线衍射图中在6.27
±
0.2
°
、10.68
±
0.2
°
、12.32
±
0.2
°
、14.99
±
0.2
°
、18.47
±
0.2
°
、20.34
±
0.2
°
、25.44
±
0.2
°
、26.26
±
0.2
°
处有特征衍射峰。
[0054]
第十七方面,本发明提供一种如第五方面所述的制备方法制得的式ⅰ所示化合物晶体n,所述式ⅰ所示化合物晶体n的x射线衍射图中在7.56
±
0.2
°
、8.78
±
0.2
°
、10.68
±
0.2
°
、12.66
±
0.2
°
、14.46
±
0.2
°
、15.98
±
0.2
°
、17.77
±
0.2
°
、18.52
±
0.2
°
处有特征衍射峰。
[0055]
相对于现有技术,本发明具有以下有益效果:
[0056]
本发明所涉及的抗hiv类药物中间体晶体的生长方法简单易操作,且制得的式ⅰ所示化合物晶体外观呈棒状型固体,有助于其纯化操作;晶体稳定,有利于减少在进行下一步反应时因在贮存过程中产生的杂质,提高合成的产率和质量;在进一步制备抗hiv类药物的反应中,该晶体还能够显著提高反应速率。以该抗hiv类药物中间体晶体为原料可进一步制备其他十三种新的式ⅰ所示化合物中间体晶体,这些晶体同样具有类似的上述有益效果。
附图说明
[0057]
图1是实施例1制得晶体的xrpd表征图;
[0058]
图2是实施例1制得晶体的dsc表征图;
[0059]
图3是实施例2制得晶体的xrpd表征图;
[0060]
图4是实施例2制得晶体的dsc表征图;
[0061]
图5是实施例3制得晶体的xrpd表征图;
[0062]
图6是实施例3制得晶体的dsc表征图;
[0063]
图7是实施例4制得晶体的xrpd表征图;
[0064]
图8是实施例4制得晶体的dsc表征图;
[0065]
图9是实施例5制得晶体的xrpd表征图;
[0066]
图10是实施例5制得晶体的dsc表征图;
[0067]
图11是实施例6制得晶体的xrpd表征图;
[0068]
图12是实施例6制得晶体的dsc表征图;
[0069]
图13是实施例7制得晶体的xrpd表征图;
[0070]
图14是实施例7制得晶体的dsc表征图;
[0071]
图15是实施例8制得晶体的xrpd表征图;
[0072]
图16是实施例8制得晶体的dsc表征图;
[0073]
图17是实施例9制得晶体的xrpd表征图;
[0074]
图18是实施例9制得晶体的dsc表征图;
[0075]
图19是实施例10制得晶体的xrpd表征图;
[0076]
图20是实施例10制得晶体的dsc表征图;
[0077]
图21是实施例11制得晶体的xrpd表征图;
[0078]
图22是实施例11制得晶体的dsc表征图;
[0079]
图23是实施例12制得晶体的xrpd表征图;
[0080]
图24是实施例12制得晶体的dsc表征图;
[0081]
图25是实施例13制得晶体的xrpd表征图;
[0082]
图26是实施例13制得晶体的dsc表征图;
[0083]
图27是实施例14制得晶体的xrpd表征图;
[0084]
图28是实施例14制得晶体的dsc表征图;
[0085]
图29是晶型b稳定性试验前后xrpd表征图;
[0086]
图30是晶型c稳定性试验前后xrpd表征图;
[0087]
图31是晶型a的dvs图;
[0088]
图32是晶型a引湿性试验前后xrpd表征图;
[0089]
图33是晶型b的dvs图;
[0090]
图34是晶型b引湿性试验前后xrpd表征图。
具体实施方式
[0091]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0092]
下述实施例中晶体所涉及的化合物均为如式ⅰ所示的化合物:
[0093][0094]
下述实施例中涉及的原料均为自制,自制方法参照专利wo2008132154。
[0095]
实施例1
[0096]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0097]
将500mg原料在60℃下溶解于6ml甲醇中,过滤。然后以0.1℃/min的速度将过滤后溶液冷却至25℃,分离得到晶体。
[0098]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:8.38
±
0.2
°
、9.44
±
0.2
°
、13.80
±
0.2
°
、16.56
±
0.2
°
、18.82
±
0.2
°
、19.08
±
0.2
°
、19.62
±
0.2
°
、23.58
±
0.2
°
,如图1所示。通过dsc进一步表征,其分别在~90℃和~174℃下具有两个吸热峰,如图2所示。
[0099]
实施例2
[0100]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0101]
将10mg实施例1晶体置于dsc样品盘中,将其使用dsc加热至120℃,即得。
[0102]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:7.09
±
0.2
°
、9.55
±
0.2
°
、10.56
±
0.2
°
、11.48
±
0.2
°
、14.08
±
0.2
°
、16.12
±
0.2
°
、17.61
±
0.2
°
,如图3所示。通过dsc进一步表征,其在~174℃下具有吸热峰,如图4所示。
[0103]
实施例3-1
[0104]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0105]
将50mg实施例2晶体悬浮于1ml正庚烷中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0106]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:6.96
±
0.2
°
、9.98
±
0.2
°
、10.40
±
0.2
°
、11.04
±
0.2
°
、13.92
±
0.2
°
、16.41
±
0.2
°
、17.97
±
0.2
°
、21.11
±
0.2
°
,如图5所示。通过dsc进一步表征,其在173℃下具有吸热峰,如图6所示。
[0107]
实施例3-2
[0108]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0109]
将50mg实施例2晶体悬浮于1ml甲苯中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0110]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:6.96
±
0.2
°
、9.98
±
0.2
°
、10.40
±
0.2
°
、11.04
±
0.2
°
、13.92
±
0.2
°
、16.41
±
0.2
°
、17.97
±
0.2
°
、21.11
±
0.2
°
,同实施例3-1。
[0111]
实施例3-3
[0112]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0113]
将50mg实施例2晶体悬浮于1ml二氯甲烷中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0114]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:6.96
±
0.2
°
、9.98
±
0.2
°
、10.40
±
0.2
°
、11.04
±
0.2
°
、13.92
±
0.2
°
、16.41
±
0.2
°
、17.97
±
0.2
°
、21.11
±
0.2
°
,同实施例3-1。
[0115]
实施例4
[0116]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0117]
将50mg实施例2晶体悬浮于1ml邻二甲苯中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0118]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:8.68
±
0.2
°
、9.89
±
0.2
°
、15.61
±
0.2
°
、16.07
±
0.2
°
、18.08
±
0.2
°
、20.82
±
0.2
°
、21.69
±
0.2
°
、26.55
±
0.2
°
,如图7所示。通过dsc进一步表征,其在109℃、114℃和173℃下具有三个吸热峰,如图8所示。
[0119]
实施例5
[0120]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0121]
将50mg实施例2晶体悬浮于1ml 2-丁醇中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0122]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:7.30
±
0.2
°
、7.57
±
0.2
°
、8.51
±
0.2
°
、14.56
±
0.2
°
、16.50
±
0.2
°
、18.38
±
0.2
°
、18.75
±
0.2
°
、21.24
±
0.2
°
,如图9所示。通过dsc进一步表征,其在104℃和173℃下具有两个吸热峰,如图10所示。
[0123]
实施例6
[0124]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0125]
将50mg实施例2晶体悬浮于1ml正丁醇中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0126]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:7.32
±
0.2
°
、7.68
±
0.2
°
、8.56
±
0.2
°
、14.64
±
0.2
°
、15.78
±
0.2
°
、16.56
±
0.2
°
、18.94
±
0.2
°
、21.36
±
0.2
°
,如图11所示。通过dsc进一步表征,其在69℃、100℃和173℃下具有三个吸热峰,如图12所示。
[0127]
实施例7
[0128]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0129]
将100mg实施例2晶体悬浮于1ml乙酸中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0130]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:7.96
±
0.2
°
、8.68
±
0.2
°
、10.53
±
0.2
°
、14.30
±
0.2
°
、15.26
±
0.2
°
、16.61
±
0.2
°
、17.40
±
0.2
°
、21.70
±
0.2
°
,如图13所示。通过dsc进一步表征,其在109℃、164℃和170℃下具有三个吸热峰,如图14所示。
[0131]
实施例8
[0132]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0133]
将100mg实施例2晶体悬浮于1ml 1,2-二氯乙烷中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0134]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:6.38
±
0.2
°
、8.76
±
0.2
°
、9.71
±
0.2
°
、15.93
±
0.2
°
、17.13
±
0.2
°
、18.89
±
0.2
°
、19.96
±
0.2
°
、22.17
±
0.2
°
,如图15所示。通过dsc进一步表征,其在123℃和173℃下具有两个吸热峰,如图16所示。
[0135]
实施例9
[0136]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0137]
将100mg实施例2晶体悬浮于1ml甲酰胺中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0138]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:6.95
±
0.2
°
、8.45
±
0.2
°
、9.56
±
0.2
°
、10.44
±
0.2
°
、13.95
±
0.2
°
、15.20
±
0.2
°
、17.00
±
0.2
°
、17.47
±
0.2
°
,如图17所示。通过dsc进一步表征,其在103℃、137℃和191℃下具有三个吸热峰,如图18所示。
[0139]
实施例10
[0140]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0141]
将100mg实施例2晶体悬浮于1ml乙醇中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0142]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:7.29
±
0.2
°
、7.55
±
0.2
°
、7.76
±
0.2
°
、8.57
±
0.2
°
、14.64
±
0.2
°
、15.77
±
0.2
°
、16.59
±
0.2
°
、19.01
±
0.2
°
,如图19所示。通过dsc进一步表征,其在75℃、106℃和173℃下具有三个吸热峰,如图20所示。
[0143]
实施例11
[0144]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0145]
将100mg实施例2晶体悬浮于1ml甲酸中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0146]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:7.52
±
0.2
°
、8.26
±
0.2
°
、9.36
±
0.2
°
、15.16
±
0.2
°
、16.64
±
0.2
°
、19.23
±
0.2
°
、19.76
±
0.2
°
、23.46
±
0.2
°
,如图21所示。通过dsc进一步表征,其在88℃、118℃、136℃和148℃下具有四个吸热峰,如图22所示。
[0147]
实施例12
[0148]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0149]
将100mg实施例2晶体悬浮于1ml 1,4-二氧嘧啶中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0150]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:6.39
±
0.2
°
、9.53
±
0.2
°
、10.00
±
0.2
°
、15.86
±
0.2
°
、16.51
±
0.2
°
、18.29
±
0.2
°
、19.30
±
0.2
°
、21.24
±
0.2
°
,如图23所示。通过dsc进一步表征,其在69℃、100℃和173℃下具有三个吸热峰,如图24所示。
[0151]
实施例13
[0152]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0153]
将500mg实施例2晶体悬浮于1ml二甲亚砜中形成混悬液,25℃下磁力搅拌3天,然后通过过滤分离残余液体,即得。
[0154]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:6.27
±
0.2
°
、10.68
±
0.2
°
、12.32
±
0.2
°
、14.99
±
0.2
°
、18.47
±
0.2
°
、20.34
±
0.2
°
、25.44
±
0.2
°
、26.26
±
0.2
°
,如图25所示。通过dsc进一步表征,其在124℃和174℃下具有两个吸热峰,如图26所示。
[0155]
实施例14
[0156]
本实施例提供一种式ⅰ所示化合物的晶体,制备方法如下:
[0157]
将500mg实施例2晶体悬浮于1ml吡啶中形成混悬液,25℃下放置3天,然后通过过滤分离残余液体,即得。
[0158]
使用xrpd(bruker d2 phaser diffractometer,2θ,cu-kα)对其进行表征,特征峰为:7.56
±
0.2
°
、8.78
±
0.2
°
、10.68
±
0.2
°
、12.66
±
0.2
°
、14.46
±
0.2
°
、15.98
±
0.2
°
、17.77
±
0.2
°
、18.52
±
0.2
°
,如图27所示。通过dsc进一步表征,其在74℃、104℃和173℃下具有三个吸热峰,如图28所示。
[0159]
评价试验:
[0160]
(1)稳定性评价
[0161]
该评价的操作方法及结果如下表所示:
[0162][0163]
其中纯度测定使用hplc,测定条件如下表所示:
[0164]
[0165][0166]
晶型b稳定性试验前后的xrpd表征图如图29所示;晶型c稳定性试验前后的xrpd表征图如图30所示。由图可知:晶型b、c在各种温度和湿度条件下均能保持很好的稳定性,纯度几乎不下降,晶型结构也能保持不变。
[0167]
(2)引湿性评价
[0168]
该评价的操作方法具体为:
[0169]
使用adventure系列动态气相吸附仪(dvs)在n2保护下收集样品的吸湿性数据,样品用量为30mg。
[0170]
测试方法如下:
[0171]
1)相对湿度增加过程:0%rh至90%rh,速率为10%rh/阶段;
[0172]
2)相对湿度降低过程:90%rh至0%rh,速率为10%rh/阶段。
[0173]
结果如图31-34所示。图31是晶型a的dvs图;图32是晶型a引湿性试验前后xrpd表征图;图33是晶型b的dvs图;图34是晶型b引湿性试验前后xrpd表征图。
[0174]
图31显示,晶型a在25℃,80%相对湿度下吸水率为0.46%;图32显示,晶体稳定,晶型没有发生变化;图33显示,晶型b在25℃,80%相对湿度下吸水率几乎为0;图34显示,晶体稳定,晶型没有发生变化。
[0175]
申请人声明,本发明通过上述实施例来说明本发明的一种抗hiv类药物中间体晶体的生长方法和所得晶体及其应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
[0176]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0177]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1