多酚类化合物及其制备方法和应用与流程
1.本发明属于药物化学领域,涉及多酚类化合物及其制备方法和在抗病毒方面的应用。
背景技术:
2.病毒在人类文明发展历程中随处可见,它给整个人类带来了巨大的危害。15世纪横扫美洲大陆的天花,18世纪肆虐全球的登革热,20世纪爆发的艾滋病,21世纪以来sars、甲型hin1、埃博拉、寨卡、流感等等,已经给全球人类的生命健康造成了巨大的危害。
3.流行性感冒(流感)是一种由流感病毒引起的病毒感染性疾病,是对人类健康威胁最大的急性呼吸道传染病之一。流行性感冒病毒(influenza virus)属于正粘病毒科(orthomyxoviridae),包括人流感病毒和动物流感病毒,人流感病毒分为甲(a)、乙(b)、丙(c)三型流感病毒。其中,甲型流感病毒的传播能力最强,发生变异也最频繁,曾引起世界范围内的多次大流行。
4.流感病毒遗传变异由环境或宿主免疫反应的选择性压力引起。在新的变异株出现的时候,及时研发并商业化生产足够有效的流感疫苗来预防疫情爆发几乎是不可能完成的任务。并且,商业化的流感疫苗的有效性不好。目前,抗流感病毒药物是治疗流行性感冒的有效措施。流感病毒m2蛋白和na蛋白在流感病毒粒子的出芽和释放过程中发挥重要作用,是抗流感病毒药物的靶点。作用于这两个靶点的临床常用抗甲型流感病毒药物有金刚烷胺、金刚乙胺等m2离子通道蛋白抑制剂和奥司他韦、扎那米韦等na神经氨酸酶抑制剂(journal of natural products,2015,78,987-995)。但是,甲型流感病毒变异快,耐药病毒株不断出现,使其在临床上的实际治疗效果越来越受限(journal of chromatography b,analytical technologies in the biomedical and life sciences,2018,15,122-130)。
5.作用于新靶点的抗流感药物成为近年来抗病毒药物研究领域的热点之一。新型抗流感病毒药物baloxavir marboxil(bxm)主要通过抑制流感病毒的cap-依赖型核酸内切酶起到抑制病毒复制的作用(scientific reports,2018,8,9633),适用于治疗12岁及以上有症状且不超过48小时的急性单纯性流感。目前已经有文献报道了bxm的耐药情况。(the annals of pharmacotherapy,2019,53,7,754-759;clinical pharmacology:advances and applications(2020),12,131-134)。
6.从天然产物中发现活性物质,并进行结构改造得到成药性更好、便于进一步研究开发的活性化合物,一直是新药发现过程中常用的研究方法。文献报道,中药五倍子提取物具有抗流感病毒作用。经研究发现,其中的单宁类物质是主要活性成分,并且该类物质广泛存在于中药民族药丹皮、诃子、白芍以及石榴果实和芒果核中。在该类物质中,人们对beta-五没食子酰葡萄糖(β-pgg)的研究比较深入,β-pgg降低病毒核蛋白(np)在质膜上积累,与病毒凝集素相互作用抑制甲型流感病毒(iav)感染,能明显降低流感病毒出芽和子代病毒释放(archives of virology,2011,156,1359-1369)。此外,β-pgg可抑制rabv病毒吸附,激
活mtor依赖性自噬信号通路灭活rabv,10mg/kg的β-pgg即可减轻病毒感染小鼠的临床症状(viruses,2018,10,4,e29673174)。β-pgg处理能够有效逆转rabv诱导的socs3/stat3信号通路,抑制病毒复制,降低il-6水平,减轻炎症反应(journal of virology,2019,93,18,e31243136)。β-pgg通过影响病毒的侵入、生物合成、释放等过程,多靶点协同作用发挥广谱抗病毒作用。对甲型流感病毒(iav)、乙肝病毒(hbv)、呼吸道合胞病毒(rsv)、单纯性疱疹病毒(hsv)的感染过程等具有明显的抑制作用(archives of virology,2011,156,1359-1369)。另外,没食子酸及其衍生物还具有广泛的药理活性,如抗氧化、抗炎、抗菌等(中成药,2017,39,6,1115-1119)。
7.但是,β-pgg主要来自于天然植物提取。由于β-pgg极性大、化学性质不稳定等原因,提取和纯化过程复杂,产品质量不稳定,产量受限。并且,pgg在体外测试中表现出一定的肝细胞增殖抑制活性。这些因素限制了pgg作为抗病毒活性化合物的进一步开发与应用。
8.在β-pgg及类似天然产物的结构基础上,进行结构优化,发现具有多靶点协同抗病毒作用,同时具有抗炎作用的广谱抗病毒活性化合物具有非常重要的意义。并且这些化合物可经过化学合成方法制备得到,样品纯度高,质量可靠,为后期研究与开发提供了充足、可靠的活性化合物来源。
技术实现要素:
9.发明目的
10.为了解决现有技术中的不足,本发明的一个目的是提供一类多酚类化合物,其具有抗病毒,特别是抗流感病毒和冠状病毒活性。
11.本发明的一个目的是提供上述化合物的制备方法。
12.本发明的再一目的是提供上述化合物在制备用于治疗或预防病毒感染的药物中的应用。
13.技术方案
14.一方面,本发明提供一类下述通式i所表示的多酚类化合物或其药学上可接受的盐或立体异构体:
[0015][0016]
其中,
[0017]
r1至r5、r
10
至r
14
各自独立地选自氢、卤素、羟基、c1~c6烷氧基、羟基c1~c6烷氧基、羟基c1~c6烷基、c1~c6烷基、卤代c1~c6烷基、氨基、被c1~c6烷基取代的氨基、被c1~c6烷酰基取代的氨基、被c1~c6烷磺酰基取代的氨基、氰基、羧基、醛基、氨基c1~c6烷
基、氰基c1~c6烷基、c1~c6烷酰基、磺酰氨基、被c1~c6烷基取代的磺酰氨基、氨基甲酰基、被c1~c6烷基取代的氨基甲酰基、羧基c1~c6烷基、c1~c6烷磺酰基、卤代c1~c6烷磺酰基、被c1~c6烷酰基取代的氨基c1~c6烷基、c1~c6烷氧基羰基;
[0018]
r6、r7为h或r6、r7与其连接的碳原子形成-c(=o);
[0019]
r8、r9为h或r8、r9与其连接的碳原子形成-c(=o);
[0020]
a为2~6价的连接基团,选自取代或未取代的下述基团:c2~c10直链或支链烷基、c2~c10直链或支链烯基、c2~c10直链或支链酯基、c3~c10环烷基、c4~c10环烯基、含有至少一个选自o、n和s的杂原子的3-15元单环杂环或双环杂环基;所述取代的取代基可以为一个或多个,各自独立地选自卤素、c1~c6烷基、卤代c1~c6烷基、c1~c6烷氧基、卤代c1~c6烷氧基、硝基、氰基、羟基、巯基、氨基、被c1~c6烷基取代的氨基、叠氮基、c1~c6烷酰基、羧基c1~c6烷基、氰基c1~c6烷基、c2~c6链烯氧基、氨基甲酰基(-conh2)、被c1~c6烷基取代的氨基甲酰基、羧基、羟基c1~c6烷基、氧代(=o)基团、硫代(=s)、氨基磺酰基(-so2nh2)、c1~c6烷硫基、c1~c6烷磺酰基、卤代c1~c6烷磺酰基、磺酸基(-so2oh)、醛基、氨基c1~c6烷基、被c1~c6烷基取代的氨基c1~c6烷基、氨基甲酰基c1~c6烷基、被c1~c6烷基取代的氨基甲酰基c1~c6烷基、c3~c10环烃基、c3~c10环烃基c1~c6烷基;
[0021]
各个x、y各自独立地选自ch2、nh、o、s或不存在,
[0022]
q和q’各自独立地为c1-c3亚烷基或不存在;
[0023]
条件是,当r6、r7与其连接的碳原子形成-c(=o),r8、r9与其连接的碳原子形成-c(=o),q和q’不存在并且a为2位被3,4,5-三羟基苯甲酰氧甲基取代的四氢吡喃基时,x、y中的至少一个不为o;
[0024]
m,n各自独立地为1~6的整数。
[0025]
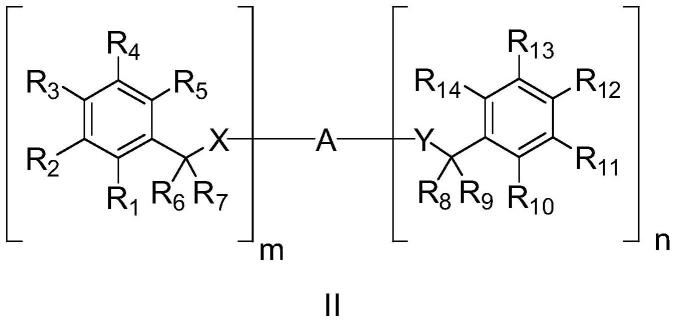
在实施方式中,所述通式i所表示的多酚类化合物为下面通式ii所示的化合物:
[0026][0027]
其中,r1至r
14
、x、y、a、m和n的定义如上所述,
[0028]
条件是,当r6、r7与其连接的碳原子形成-c(=o),r8、r9与其连接的碳原子形成-c(=o),且a为2位被3,4,5-三羟基苯甲酰氧甲基取代的四氢吡喃基时,x、y中的至少一个不为o。
[0029]
在一些实施方式中,r1至r5、r
10
至r
14
各自独立地选自氢、卤素、羟基、c1~c3烷氧基、羟基c1~c3烷氧基、羟基c1~c3烷基、c1~c3烷基、卤代c1~c3烷基、氨基、被c1~c3烷基取代的氨基、被c1~c3烷酰基取代的氨基、被c1~c6烷磺酰基取代的氨基、氰基、羧基、c1~c3烷基取代的羧基、醛基、c1~c3烷酰基、磺酰氨基、被c1~c3烷基取代的磺酰氨基、氨基
甲酰基、被c1~c3烷基取代的氨基甲酰基、c1~c3烷磺酰基、卤代c1~c3烷磺酰基、c1~c3烷氧基羰基。
[0030]
在一些实施方式中,a选自取代或未取代的下述基团:c2~c10直链或支链烷基,c2~c10直链或支链烯基,c2~c10直链或支链酯基,c3~c10环烷基,c4~c10环烯基,含有至少一个选自o、n和s的杂原子的3-12元单环杂环或双环杂环基;优选地,a为被3,4,5-三羟基苯甲酰氧甲基取代的四氢吡喃基。所述取代的取代基定义如上所述,特别地,所述取代的取代基可以为一个或多个,各自独立地选自卤素、c1~c3烷基、卤代c1~c3烷基、c1~c3烷氧基、卤代c1~c3烷氧基、硝基、氰基、羟基、巯基、氨基、被c1~c3烷基取代的氨基、叠氮基、c1~c3烷酰基、羧基c1~c3烷基、氰基c1~c3烷基、氨基甲酰基(-conh2)、被c1~c3烷基取代的氨基甲酰基、c1~c3烷基取代的羧基、羟基c1~c3烷基、氧代(=o)基团、硫代(=s)、氨基磺酰基(-so2nh2)、c1~c3烷硫基、c1~c3烷磺酰基、卤代c1~c3烷磺酰基、磺酸基(-so2oh)、醛基、氨基c1~c3烷基。
[0031]
特别地,a为衍生自含有羟基和/或氨基的化合物的2~6价的连接基团,例如,所述含有羟基和/或氨基的化合物选自下列各项中的任一种:d-(+)-葡萄糖胺、d-(+)-半乳糖胺、n-乙酰基-d-葡萄糖胺、n-乙酰基-d-半乳糖胺、抗坏血酸、l-半胱氨酸乙酯、3,7-二氮杂双环[3.3.0]辛烷、绿原酸、丝氨醇、1,3-二氨基-2-丙醇、葡甲胺。
[0032]
在一些实施方式中,r2至r4、r
11
至r
13
为羟基,r1、r5、r
10
和r
14
为氢。
[0033]
在一些实施方式中,所述通式i所表示的多酚类化合物为如下的通式ia、ib、ic所示的化合物:
[0034][0035]
其中,
[0036]
在ia中,r1至r5、a的定义同上文所述;x、y各自独立地选自ch2、nh、o或s,条件是,a为2位被3,4,5-三羟基苯甲酰氧甲基取代的四氢吡喃基时,x、y中的至少一个不为o;
[0037]
在ib中,r1至r5、a、x、y的定义同上文所述;
[0038]
在ic中,r1至r5、a、x、y的定义同上文所述;
[0039]
在以上ia、ib和ic中,m和n的定义分别同上文所述。
[0040]
在一些实施方式中,所述化合物为具有以下结构的化合物之一:
[0041][0042][0043]
本发明中,
[0044]
卤素指的是氟、氯、溴和碘;
[0045]
羟基指的是-oh;
[0046]
c1~c6烷基指的是具有1~6个碳原子的烷基,例如甲基、乙基、正丙基、异丙基、正
丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、正己基等;c2~c10直链或支链烷基的含义以此类推。
[0047]
c1~c6烷氧基指的是上述c1~c6烷基末端连接一个氧所得的基团,即c1~c6烷基-o-;
[0048]
羟基c1~c6烷氧基指的是上述c1~c6烷氧基的一个或多个氢被羟基取代所得的基团;
[0049]
羟基c1~c6烷基指的是上述c1~c6烷基的一个或多个氢被羟基取代所得的基团;
[0050]
卤代c1~c6烷基指的是上述c1~c6烷基的一个或多个氢被卤素取代所得的基团;
[0051]
氨基指的是-nh2;
[0052]
被c1~c6烷基取代的氨基指的是上述-nh2的一个或两个氢被c1~c6烷基取代所得的基团,即c1~c6烷基-nh-或(c1~c6烷基)
2-n-;
[0053]
c1~c6烷酰基指的是在上述c1~c6烷基末端连接一个羰基所得的基团,即c1~c6烷基-c(=o)-;
[0054]
c1~c6烷磺酰基指的是在上述c1~c6烷基末端连接一个磺酰基所得的基团,即c1~c6烷基-s(=o)
2-;
[0055]
卤代c1~c6烷磺酰基指的是在上述c1~c6烷磺酰基的c1~c6烷基上的一个或多个氢被卤素取代所得的基团;
[0056]
被c1~c6烷酰基取代的氨基指的是上述-nh2的一个氢被c1~c6烷酰基取代所得的基团,即c1~c6烷基-c(=o)-nh-;
[0057]
被c1~c6烷磺酰基取代的氨基指的是上述-nh2的一个氢被c1~c6烷磺酰基取代所得的基团,即c1~c6烷基-s(=o)
2-nh-;
[0058]
氰基指的是-cn;
[0059]
羧基指的是-c(=o)oh;
[0060]
醛基指的是-c(=o)h;
[0061]
氨基c1~c6烷基指的是上述c1~c6烷基的一个或多个氢被氨基取代所得的基团;
[0062]
被c1~c6烷基取代的氨基c1~c6烷基指的是上述氨基c1~c6烷基中氨基的一个或多个氢被c1~c6烷基取代所得的基团;
[0063]
氰基c1~c6烷基指的是上述c1~c6烷基的一个或多个氢被氰基取代所得的基团;
[0064]
氨基磺酰基指的是-s(=o)2nh2;
[0065]
磺酰氨基指的是r-s(=o)
2-nh-,其中r为c1-c10烷基或c6-c10芳基(例如苯基、萘基);
[0066]
被c1~c6烷基取代的磺酰氨基指的是c1~c6烷基-s(=o)
2-nh-;
[0067]
氨基甲酰基指的是h2n-c(=o)-,即(-conh2);
[0068]
被c1~c6烷基取代的氨基甲酰基指的是h2n-c(=o)-中氨基上的氢被一个或两个c1~c6烷基取代所得的基团,即c1~c6烷基-nh-c(=o)-或(c1~c6烷基)
2-n-c(=o)-;
[0069]
氨基甲酰基c1~c6烷基指的是上述c1~c6烷基的一个或多个氢被氨基甲酰基取代所得的基团;
[0070]
被c1~c6烷基取代的氨基甲酰基c1~c6烷基指的是上述氨基甲酰基c1~c6烷基中氨基的一个或多个氢被c1~c6烷基取代所得的基团;
[0071]
羧基c1~c6烷基指的是上述c1~c6烷基的一个或多个氢被羧基取代所得的基团;
[0072]
被c1~c6烷酰基取代的氨基c1~c6烷基指的是上述氨基c1~c6烷基中氨基上的一个或多个氢被c1~c6烷酰基取代所得的基团;
[0073]
c1~c6烷氧基羰基指的是上述c1~c6烷氧基末端连接一个羰基所得的基团,即c1~c6烷基-o-c(=o)-;
[0074]
c2~c10直链或支链烯基指的是主链中含有至少一个碳碳双键的具有2至10个碳原子的烯基;
[0075]
c2~c10直链或支链酯基指的是c2~c10直链或支链烷基主链中含有酯基基团,即r
’‑
c(=o)-o-r”,r’和r”分别独立地表示c1~c10直链或支链烷基,或者r’和r”之一不存在;
[0076]
c3~c10环烷基指的是环上具有3至10个碳原子的环状烷基,例如环丙基、环丁基、环戊基、环己基等;
[0077]
c4~c10环烯基指的是环上具有至少一个碳碳双键的具有4至10个碳原子的环状烯基,例如环丁烯基、环戊烯基、环己烯基等;
[0078]
3-15元单环杂环或双环杂环基指的是环上具有3~15个原子的单环或双环基,其中包含至少一个选自o、n和s的杂原子。
[0079]
巯基指的是-sh;
[0080]
叠氮基指的是-n=n=n;
[0081]
c2~c6链烯氧基指的是c2~c6直链或支链烯基末端连接一个氧所得的基团,即c2~c6直链或支链烯基-o-;
[0082]
c1~c6烷基羧基指的是c2~c6烷基末端连接一个羧基基团,即c2~c6烷基-c(=o)-oh;
[0083]
c1~c6烷硫基指的是上述c1~c6烷基末端连接一个硫所得的基团,即c1~c6烷基-s-;
[0084]
磺酸基指的是-so2oh;
[0085]
c3~c10环烃基指的是环上具有3至10个碳原子的环烷基或环烯基,即c3~c10环烃基包括c3~c10环烷基和c3~c10环烯基;
[0086]
c3~c10环烃基c1~c6烷基指的是上述c1~c6烷基的一个或多个氢被c3~c10环烃基取代所得的基团。
[0087]
本发明还提供了通式i化合物的制备方法。
[0088]
其中,通式ia化合物可以通过如下方法一或方法二制备得到:
[0089]
方法一包括以下步骤:
[0090]
步骤a:以化合物iii和iv为原料,在缩合剂存在的条件下得到缩合产物iia;以及
[0091]
步骤b:化合物iia在脱保护条件下反应得到化合物ia,以上反应如以下反应式1所示:
[0092]
反应式1:
[0093][0094]
其中,ia中r1至r5、a同上文所述;x、y各自独立地选自ch2、nh、o或s,条件是x、y中的至少一个不为o,r1’
至r5’
分别为r1至r5的带保护基团的取代基,所述保护基团为本领域中常见的保护基团,例如为苄基;
[0095]
在步骤a中使用的缩合剂可选自:1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edci)、二环己基碳二亚胺(dcc)、1-羟基苯并三唑(hobt)中一种或几种的组合,或可以使用本领域中类似机理的缩合试剂。
[0096]
在步骤b中,所述脱保护反应条件是指:含有氢气或其他还原剂及催化剂的混合体系、含有路易斯酸或路易斯碱的混合体系;
[0097]
方法二包括以下步骤:
[0098]
步骤c:以化合物iii为原料,在草酰氯或二氯亚砜存在的条件下制备化合物iiia;
[0099]
步骤d:所得化合物iiia与化合物iv反应得到化合物iia;以及
[0100]
步骤e:化合物iia在脱保护条件下反应得到化合物ia,
[0101]
如反应式2所示:
[0102]
反应式2:
[0103][0104]
其中,r1至r5、a同上文所述;x、y各自独立地选自ch2、nh、o或s,条件是x、y中的至少一个不为o,r1’
至r5’
分别为r1至r5的带保护基团的取代基;
[0105]
z为离去基团,可选自卤素、对硝基苯基氧基、五氟苯基氧基、n-丁二酰亚胺基氧基、n-邻苯二酰亚胺基氧基、乙酰氧基或苯甲酰氧基;
[0106]
在步骤e中,所述脱保护反应条件是指:含有氢气或其他还原剂及催化剂的混合体系、含有路易斯酸或路易斯碱的混合体系。
[0107]
通式ib化合物可通过如下方法制备得到,所述方法包括以下步骤:
[0108]
步骤f:以化合物iiib和iv为原料,在碱存在的条件下发生烃基化反应,得到化合物iib;
[0109]
步骤g:化合物iib在脱保护条件下反应得到化合物ib,如反应式3所示:
[0110]
反应式3:
[0111][0112]
其中,以上反应式中,r1至r5、a、x、y、m、n的定义上文所述,r1’
至r5’
分别为r1至r5的带保护基团的取代基。
[0113]
l为离去基团,可选自卤素、甲磺酸酯基、三氟甲磺酸酯基、对甲基苯磺酸酯基;
[0114]
在步骤f中,所述碱为有机碱或无机碱,可选自碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、氢氧化锂、氢化钠、氢化钙、甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾、吡啶、三乙胺、二异丙基乙胺、n,n-二甲基氨基吡啶中的一种或几种的混合物。
[0115]
在步骤g中,脱保护反应条件是指:含有氢气或其他还原剂及催化剂的混合体系、含有路易斯酸或路易斯碱的混合体系。
[0116]
通式ic化合物的制备方法同以上通式ib化合物的制备方法,不同之处仅在于用以下反应物iiic替代反应物iiib:
[0117][0118]
在通式iiic中,r1’
至r5’
以及l的定义分别同上文所定义。
[0119]
另一方面,本发明还提供一种药物组合物,其包含治疗有效量的上述通式i的化合物中的一种或几种,和任选的可药用载体。
[0120]
另一方面,本发明提供所述化合物或所述药物组合物在制备抗病毒药物中的用途。
[0121]
在一些实施方式中,所述病毒为dna病毒、rna病毒中的至少一种。
[0122]
在一些实施方式中,所述dna病毒为乙型肝炎病毒、人乳头瘤病毒、疱疹病毒、噬菌体病毒;所述rna病毒为流感病毒、禽流感病毒、寨卡病毒、登革热病毒、呼吸道合胞病毒、sars病毒、新型冠状病毒(2019-ncov)、艾滋病病毒、埃博拉病毒、丙型肝炎病毒、乙型脑炎病毒、鼻病毒、脊髓灰质炎病毒、柯萨奇病毒、轮状病毒、烟草花叶病毒、噬菌体病毒、mers病毒、马尔堡病毒、肠道病毒。
[0123]
在一些实施方式中,所述流感病毒为甲型、乙型和丙型流感病毒中的至少一种。
[0124]
在一些实施方式中,所述流感病毒为甲型h1n1流感病毒。
[0125]
有益效果
[0126]
本发明化合物具有以下有益效果:结构新颖、具有显著抗病毒活性。
具体实施方式
[0127]
为了进一步理解本发明,下面结合实施例对本发明作进一步的说明,这些实施例描述只是为进一步详细说明本发明的特征,而不是对本发明范围或本发明权利要求范围的限制。在本领域内技术人员对发明所做的简单替换或改进等均应包含在本发明所保护的技术方案之内。
[0128]
制备例1:化合物i1的合成
[0129][0130]
将20.0g没食子酸和200ml甲醇加入到500ml三口瓶中,冰水浴条件下,将15ml浓硫酸缓慢滴加至反应液中,逐渐升温至70℃,反应6h。减压蒸馏除去甲醇,加入300ml乙酸乙酯和50ml水搅拌5min后分层,有机相加入50ml饱和食盐水搅拌5min后分层,无水硫酸钠干燥有机相1h。最后通过柱层析法(sio2,二氯甲烷:甲醇=30:1)进行纯化,得到19.3g白色固体产物,收率89.3%。将17.0g原料、300ml丙酮和52.0g碳酸钾加入到1000ml三口瓶中,25℃条件下,将57.0g溴苄滴加至反应液中,逐渐升温至70℃,反应12h。
[0131]
将反应液降温至25℃,过滤反应液,减压蒸馏除去丙酮,加入500ml乙酸乙酯和100ml水搅拌5min后分层,有机相加入100ml饱和食盐水搅拌5min后分层,无水硫酸钠干燥有机相1h。过滤有机相,减压蒸馏除去乙酸乙酯,加入200ml石油醚,50℃打浆1h后,过滤固体,烘干后得到35.5g白色固体产物,收率84.5%。将35.0g原料、100ml甲醇和200ml二氧六环加入到1000ml三口瓶中,25℃条件下,将60ml 5m氢氧化钠水溶液滴加至反应液中,逐渐升温至90℃,反应1h。减压蒸馏反应液至约50ml,加入2n盐酸溶液调节ph≤4,大量固体析出,过滤固体,烘干后得到29.0g白色固体产物i1,收率88.4%。1h nmr(dmso,500mhz):12.95(s,4h),7.48(m,4h),7.43-7.34(m,11h),7.30(m,2h),5.20(s,4h),5.06(s,2h).esi-ms m/z 463.2(m+na)
+
.
[0132]
制备例2:化合物i2的合成
[0133][0134]
将250mg i1、165mg n,n
′‑
二环己基碳二亚胺(dcc)、100mg 4-二甲氨基吡啶(dmap)和10ml二氯甲烷加入到50ml三口瓶中,25℃条件下搅拌10min后将15mg乙二醇加入至反应液中,反应20h。
[0135]
加入5ml水分层,浓缩有机相通过柱层析法(sio2,石油醚:乙酸乙酯=3:1)进行纯化,得到180mg白色固体。
[0136]
将150mg上述固体、3ml甲醇和3ml四氢呋喃加入到10ml单口烧瓶中,加入75mg 10%pd/c,氢气置换三次,25℃条件下反应15h。通过硅藻土过滤反应液,浓缩反应液后通过反相柱(c18,水:甲醇=4:1)进行纯化,得到35mg白色固体产物i2,两步收率39.8%。1h nmr(meod,500mhz):7.07(s,4h),4.53(s,4h).esi-ms m/z 365.0(m-h)-,730.5(2m-h)-.
[0137]
制备例3:化合物i3的合成
[0138]
[0139]
按照制备例2中相同的方法,不同之处在于以甘油代替乙二醇,两步收率50.0%。1h nmr(dmso,500mhz):6.96(s,2h),6.95(s,4h),5.56(ddd,1h),4.57(dd,2h),4.48(dd,2h).esi-ms m/z 547.2(m-h)-.
[0140]
制备例4:化合物i4的合成
[0141][0142]
按照制备例2中相同的方法,不同之处在于以4-苄氧基苯甲酸代替i1和以甘油代替乙二醇,两步收率34.3%。1h nmr(dmso,500mhz):7.80(m,4h),6.83(m,4h),5.64(m,1h),4.63(dd,2h),4.56(dd,2h).esi-ms m/z451.1(m-h)-.
[0143]
制备例5:化合物i5的合成
[0144][0145]
按照制备例2中相同的方法,不同之处在于以d-木糖代替乙二醇,两步收率14.3%。1h nmr(dmso,500mhz):6.92(s,2h),6.88(s,2h),6.84(d,4h),6.19(d,1h),5.86(t,1h),5.45(dd,1h),5.28(td,1h),4.21(dd,1h),3.93(t,1h).esi-ms m/z 775.9(m+nh4)
+
.
[0146]
以上制备例2-5合成的化合物i2-i5将在以下药理实验中用作参照化合物。
[0147]
实施例1:化合物ia1的合成
[0148][0149]
将1500mg i1、806mg dcc、980mg dmap和15ml二氯甲烷(dcm)加入到50ml三口瓶中,25℃条件下搅拌10min后将100mg d-(+)-葡萄糖胺盐酸盐加入至反应液中,逐渐升温至45℃,反应20h。过滤反应液,将有机相通过柱层析法(sio2,二氯甲烷:甲苯:乙酸乙酯=30:10:2)进行纯化,得到500mg白色固体。
[0150]
将400mg上述固体、4ml甲醇和4ml四氢呋喃(thf)加入到25ml单口烧瓶中,加入200mg 10%pd/c,氢气置换三次,25℃条件下反应15h。通过硅藻土过滤反应液,浓缩反应液
后通过反相柱(c18,水:甲醇=2:1)进行纯化,得到75mg晶状固体产物ia1,两步收率21.5%。1h nmr(dmso,500mhz):8.39(d,1h),7.21(s,2h),6.98(s,2h),6.88(s,2h),6.82(s,2h),6.71(s,2h),6.39(d,1h),5.87(m,1h),5.47(t,1h),4.70(ddd,1h),4.38(m,1h),4.26(t,2h).esi-ms m/z 938.2(m-h)-.
[0151]
实施例2:化合物ia2的合成
[0152][0153]
按照实施例1中相同的方法,不同之处在于以d-(+)-半乳糖胺盐酸盐代替d-(+)-葡萄糖胺盐酸盐,两步收率12.7%。1h nmr(dmso,500mhz,包含α/β两种构型):8.64(d,1h),8.15(d,1.3h),7.16(s,2h),6.99(m,7h),6.92(s,3h),6.85(s,2h),6.81(d,4h),6.78(s,3h),6.66(s,3h),6.40(d,1h),6.13(dd,1h),6.05(d,1.3h),5.70(d,1.3h),5.53(m,2.3h),4.97(td,1h),4.79(q,1.3h),4.59(d,1h),4.48(m,3.3h),4.23(ddd,2.6h).esi-ms m/z 938.2(m-h)-.
[0154]
实施例3:化合物ia3的合成
[0155][0156]
按照实施例1中相同的方法,不同之处在于以n-乙酰基-d-葡萄糖胺代替d-(+)-葡萄糖胺盐酸盐,两步收率26.6%。1h nmr(ch3od,500mhz,包含α/β两种构型):7.26(s,2h),7.10-7.08(m,3h),6.99-6.96(m,6h),6.44(d,1h),5.98(d,0.4h),5.81(dd,1h),5.65-5.60(m,1.4h),5.53(t,0.4h),4.73(dd,1h),4.55-4.52(m,0.4h),4.49(ddd,1.4h),4.43(dd,1h),4.35(ddd,1.4h),4.27(ddd,0.4h),1.86(s,3h),1.83(s,1.3h).esi-ms m/z 827.9(m-h)-.
[0157]
实施例4:化合物ia4的合成
[0158][0159]
按照实施例1中相同的方法,不同之处在于以n-乙酰基-d-半乳糖胺代替d-(+)-葡萄糖胺盐酸盐,两步收率16.0%。1h nmr(ch3od,500mhz):7.20(s,2h),7.11(s,2h),7.20(s,2h),6.98(s,2h),6.94(s,2h),6.57(d,1h),5.99(dd,1h),5.60(dd,1h),4.99(dd,1h),4.71(m,1h),4.44(dd,1h),4.21(dd,1h),1.87(s,3h).esi-ms m/z 827.9(m-h)-.
[0160]
实施例5:化合物ia5的合成
[0161][0162]
将200mg i1、5ml甲苯加入到10ml三口瓶中,25℃条件下滴加0.1ml草酰氯,搅拌10min后升温至50℃,反应1h。浓缩反应液,将2ml甲苯加入至反应液中,升温至70℃,搅拌1h后,转移上清液至10ml单口瓶中,将3ml环己烷滴加至反应液中,室温搅拌30min后过滤酰氯固体,隔膜泵拉干备用。将25mg抗坏血酸、5ml二氯甲烷、0.2ml吡啶和1.0ml dmf加入到25ml三口瓶中,25℃条件下将酰氯固体一次性加入反应液中,反应15h。浓缩反应液,通过柱层析法(sio2,二氯甲烷:甲醇=50:1)进行纯化,得到120mg白色固体。
[0163]
将120mg上述固体、3ml甲醇和3ml四氢呋喃加入到25ml单口烧瓶中,加入100mg 10%pd/c,氢气置换三次,25℃条件下反应12h。通过硅藻土过滤反应液,浓缩反应液后通过反相柱(c18,水:甲醇=2:1)进行纯化,得到30mg白色固体产物ia5,两步收率33.3%。1h nmr(ch3od,500mhz):7.15(s,2h),7.09(s,4h),5.80(d,1h),5.11(d,1h),4.69(dd,1h),4.59(dd,1h).esi-ms m/z 630.8(m-h)-.
[0164]
实施例6:化合物ia6的合成
[0165][0166]
将50mg l-半胱氨酸乙酯盐酸盐、5ml二氯甲烷、0.2ml吡啶和1.0ml dmf加入到25ml三口瓶中,25℃条件下将300mg三苄基没食子酰氯固体一次性加入反应液中,反应15h。
浓缩反应液,通过柱层析法(sio2,石油醚:二氯甲烷=1:2)进行纯化,得到220mg白色固体。
[0167]
将150mg上述固体和5ml二氯甲烷加入到10ml三口烧瓶中,氮气置换三次,-25℃条件下,缓慢滴加1.5ml 1m三氯化硼,逐渐升温至25℃条件下反应3h。0℃条件下将10ml甲醇缓慢滴加至反应液中,逐渐升温至25℃反应1h。浓缩反应液后通过反相柱(c18,水:甲醇=2:1)进行纯化,得到45mg白色固体产物ia6,两步收率54.9%。1h nmr(ch3od,500mhz):7.15(s,2h),7.09(s,4h),5.80(d,1h),5.11(d,1h),4.69(dd,1h),4.59(dd,1h).esi-ms m/z 451.9(m-h)-.
[0168]
实施例7:化合物ia7的合成
[0169][0170]
将1100mg i1、150mg 3,7-二氮杂双环[3.3.0]辛烷二盐酸盐、824mg dcc、882mg dmap和10ml二氯甲烷加入到50ml三口瓶中,25℃条件下反应15h。过滤反应液,将有机相通过柱层析法(sio2,二氯甲烷:甲醇=50:1)进行纯化,得到600mg白色固体。
[0171]
将300mg上述固体、5ml甲醇和5ml四氢呋喃加入到25ml单口烧瓶中,加入200mg 10%pd/c,氢气置换三次,25℃条件下反应15h。通过硅藻土过滤反应液,浓缩反应液后通过反相柱(c18,水:甲醇=1:1)进行纯化,得到70mg白色固体产物ia7,两步收率25.1%。1h nmr(dmso,500mhz):6.48(s,4h),3.66(brs,4h),3.31(m,4h),2.87(s,2h).esi-ms m/z415.0(m-h)-.
[0172]
实施例8:化合物ia8的合成
[0173][0174]
将92mg绿原酸、5ml二氯甲烷、0.2ml吡啶和1.0ml dmf加入到25ml三口瓶中,25℃条件下将250mg三苄基没食子酰氯固体一次性加入反应液中,反应15h。浓缩反应液,通过柱层析法(sio2,二氯甲烷:甲醇=15:1)进行纯化,得到80mg白色固体。
[0175]
将80mg上述固体和5ml二氯甲烷加入到10ml三口烧瓶中,氮气置换三次,-25℃条件下,缓慢滴加0.7ml 1m三氯化硼,逐渐升温至25℃条件下反应3h。0℃条件下将10ml甲醇缓慢滴加至反应液中,逐渐升温至25℃反应1h。浓缩反应液后通过反相柱(c18,水:甲醇=1:1)进行纯化,得到22mg白色固体产物ia8,两步收率12.9%。1h nmr(dmso,500mhz):7.78(d,1h),7.00(m,4h),6.52(d,1h),5.52(t,1h),4.34(d,1h),3.84(dd,1h),2.79(t,2h),2.31(m,2h).esi-ms m/z 656.9(m-h)-.
[0176]
实施例9:化合物ia9的合成
[0177][0178]
将800mg 3,4-二苄氧基苯甲酸、80mg丝氨醇、1063mg edci、752mg dmap和10ml二氯甲烷加入到50ml三口瓶中,45℃条件下反应15h。将反应液降温至25℃,加入10ml 1n hcl分层,浓缩有机相通过柱层析法(sio2,石油醚:乙酸乙酯=3:1)进行纯化,得到500mg白色固体。
[0179]
将250mg上述固体、5ml甲醇加入到10ml单口烧瓶中,加入200mg10%pd/c,氢气置换三次,25℃条件下反应15h。通过硅藻土过滤反应液,浓缩反应液后通过反相柱(c18,水:甲醇=3:1)进行纯化,得到100mg白色固体产物ia9,两步收率45.5%。1h nmr(dmso,500mhz):8.32(d,1h),7.38(d,2h),7.35(dd,2h),7.30(d,1h),7.21(dd,1h),6.80(d,2h),6.76(d,1h),4.63(m,1h),4.39(ddt,4h).esi-ms m/z 497.9(m-h)-.
[0180]
实施例10:化合物ia10的合成
[0181][0182]
按照实施例1中相同的方法,不同之处在于以3-甲氨基-1,2-丙二醇代替d-(+)-葡萄糖胺盐酸盐,两步收率18.2%。1h nmr(ch3od,500mhz):7.11(s,4h),6.40(s,2h),5.73(s,1h),4.61(s,1h),4.46(s,1h),4.05(s,1h),3.87(d,1h),3.13(s,3h).esi-ms m/z 560.0(m-h)-.
[0183]
实施例11:化合物ia11的合成
[0184][0185]
按照实施例1中相同的方法,不同之处在于以1,3-二氨基-2-丙醇代替d-(+)-葡萄糖胺盐酸盐,两步收率12.5%。1h nmr(dmso,500mhz):6.96(s,2h),6.82(s,4h),5.11(p,1h),3.47(m,4h).esi-ms m/z 545.1(m-h)-.
[0186]
实施例12:化合物ia12的合成
[0187][0188]
按照实施例1中相同的方法,不同之处在于以葡甲胺代替d-(+)-葡萄糖胺盐酸盐,两步收率16.0%。1h nmr(dmso,500mhz):6.92(m,10h),6.14(s,2h),5.86(s,1h),5.77(s,2h),5.45(s,1h),4.69(d,1h),4.42(s,1h),3.50(d,1h),2.84(s,3h).esi-ms m/z 1105.8(m-h)-,552.6(m/2-h)-.
[0189]
实施例13:化合物ib1的合成
[0190][0191]
将100mg d-葡萄糖、10ml无水dmf加入到50ml三口瓶中,氮气置换三次,0℃条件下将132mg氢化钠(60%)缓慢加入反应液中,0℃反应1h。将1760mg 3,4,5-三苄氧基溴苄加入至反应液中,缓慢升温至25℃,反应12h。
[0192]
将50ml乙酸乙酯和30ml水加入反应液中,搅拌10min,静置分层。浓缩有机相通过柱层析法(sio2,石油醚:乙酸乙酯=3:2)进行纯化,得到350mg白色固体。
[0193]
将300mg上述固体和5ml二氯甲烷加入到10ml三口烧瓶中,氮气置换三次,-25℃条件下,缓慢滴加2.5ml 1m三氯化硼,逐渐升温至25℃条件下反应1h。0℃条件下将10ml甲醇缓慢滴加至反应液中,逐渐升温至25℃反应1h。浓缩反应液后通过反相柱(c18,水:甲醇=2:1)进行纯化,得到50mg白色固体产物ib1,两步收率11.9%。1h nmr(dmso,500mhz):7.03(s,2h),6.66(s,2h),6.31(s,2h),6.20(s,2h),6.09(s,2h),5.56(m,1h),5.11(m,1h),4.83-4.51(m,12h),4.13(m,1h),4.08(t,2h).esi-ms m/z 869.62(m-h)-.
[0194]
实施例14:化合物ib2的合成
[0195][0196]
按照实施例13中相同的方法,不同之处在于以d-(+)-葡萄糖胺代替d-葡萄糖,两步收率10.2%.1h nmr(dmso,500mhz):7.38(s,1h),6.99(s,2h),6.72(s,2h),6.26(s,2h),6.14(s,2h),6.03(s,2h),5.73(d,1h),5.22(m,1h),4.61-4.36(m,12h),4.23(m,1h),4.05
(t,2h).esi-ms m/z 868.37(m-h)-.
[0197]
实施例15:化合物ic1的合成
[0198][0199]
按照实施例13中相同的方法,不同之处在于以3,4,5-三苄氧基-2-溴代苯乙酮代替3,4,5-三苄氧基溴苄,两步收率15.8%.1h nmr(dmso,500mhz):7.11(s,2h),6.96(s,2h),6.53(s,2h),6.33(s,2h),6.19(s,2h),5.82(d,1h),5.29(m,1h),4.93-4.56(m,12h),4.43(dd,1h),4.14(t,2h).esi-ms m/z1009.76(m-h)-.
[0200]
药理实验
[0201]
细胞病变效应(cpe)实验
[0202]
将实验细胞以一定的细胞密度(见表1)接种到96孔细胞培养板中、于5%co2、37℃培养箱中培养过夜。第二天加入化合物(8个浓度点、双复孔)和病毒。依所测病毒不同而异,细胞在5%co2、33℃或37℃条件下于培养箱中培养3-7天(表1),直至无化合物的病毒感染对照孔内细胞病变达80-95%。然后用celltiter-glo或细胞增殖及毒性检测试剂盒(cells counting kit 8,cck-8)检测每孔细胞活力。如含化合物孔的细胞活力较病毒感染对照孔高,即cpe减弱,则表明化合物对所测病毒有抑制作用。细胞毒性实验与相应的抗病毒实验相同,但无病毒感染。
[0203]
表1.所用病毒试验方法
[0204][0205]
化合物的抗病毒活性由化合物对病毒引起的细胞病毒效应的抑制率(%)表示,见表2。计算公式如下:抑制率(%)=(测试孔读值-病毒对照平均值)/(细胞对照平均值-病毒对照平均值)
×
100
[0206]
表2.不同浓度抑制率
[0207]
测试化合物5μm抑制率(%)10μm抑制率(%)i2***i3n/a**i4**i5***
ia1******ia2******ia3*****ia4********ia5**ia6***ia7**ia8n/an/aia9***ia10***ia11****ia12n/a*
[0208]
说明:
[0209]
n/a:无抑制
[0210]
*:1-25
[0211]
**:26-50
[0212]
***:51-75
[0213]
****:76-100
[0214]
从以上表2中的数据可以看出,本技术实施例的化合物,特别是实施例1-4的化合物在流感病毒抑制活性方面显著优于制备例2-5的化合物,因此,表现出了较高的流感病毒抑制活性,具有可被开发成抗病毒药物的潜力。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1