无色透明树脂薄膜的制造方法与流程
1.本发明涉及无色透明树脂薄膜的制造方法。
背景技术:
2.聚酰亚胺树脂由于具有优异的机械特性和耐热性,因此,研究了其在电气部件或电子部件等领域中的各种用途。例如,出于器件的轻量化、柔性化的目的,期望将液晶显示器、有机el显示器等图像显示装置中使用的玻璃基板替换为塑料基板,也进行了适合作为该塑料材料的聚酰亚胺树脂的研究。
3.通用聚酰亚胺薄膜通常如下进行溶液流延制膜:使含有聚酰胺酸的有机溶剂溶液在带或鼓上流延,将聚酰胺酸进行加热等酰亚胺化反应,进行溶液流延制膜。
4.已知通用聚酰亚胺具有高的耐热性。通用聚酰亚胺由芳香族四羧酸酐和芳香族二胺得到,由于分子的刚性、共振稳定化、强的化学键合而具有优异的耐热性、耐化学药品性、机械物性、电气特性,因此,在成型材料、复合材料、电气部件或电子部件等领域中被广泛使用。
5.然而,对于由上述芳香族原料合成的聚酰亚胺类,由于源自分子内或分子间的电子转移络合物产生的吸收而在成型为薄膜后着色为黄色至褐色,因此,不适于平板显示器、移动电话设备等的基板材料、光纤、光波导、光传感器、光学用粘接剂等光学用途。
6.因此,为了发挥聚酰亚胺的高耐热性的特征、且为了改善透明性,逐渐开发了在重复单元中导入了全氟烷基的氟化聚酰亚胺、使用了1,2,4,5
‑
环己烷四羧酸酐等脂环式原料的脂环式聚酰亚胺。
7.专利文献1中公开了一种由含有聚酰亚胺系高分子、以及γ
‑
丁内酯和n,n
‑
二甲基乙酰胺的液体制造剥离性和外观优异的聚酰亚胺系薄膜的制造方法。
8.专利文献2中公开了如下光学薄膜的制造方法:其通过在以可溶性聚酰亚胺树脂为主成分、以二氯甲烷为溶剂的液体中含有醇系溶剂,从而减少了鳞片状的不均。
9.现有技术文献
10.专利文献
11.专利文献1:日本特开2017
‑
25204号公报
12.专利文献2:日本特开2017
‑
187617号公报
技术实现要素:
13.发明要解决的问题
14.然而,专利文献1的方法中,使用γ
‑
丁内酯、n,n
‑
二甲基乙酰胺那样的高沸点的有机溶剂的情况下,为了去除溶剂,必须升高干燥工序中的温度,存在由于着色而透明性降低的问题。
15.另外,专利文献2的方法中,二氯甲烷那样的低沸点溶剂的情况下,溶剂会立即挥发,因此,通过流延法取向的聚酰亚胺被固定,存在产生光学各向异性的问题。
16.本发明要解决的课题在于,提供:无高温干燥下的着色、能实现良好的光学各向同性的无色透明树脂薄膜的制造方法。
17.用于解决问题的方案
18.本发明人等发现:以溶液流延法制造薄膜时,通过树脂的有机溶剂溶液中的有机溶剂中含有低沸点溶剂作为主成分、且含有高沸点溶剂,从而可以解决上述课题,至此完成了的本发明。
19.即,本发明涉及一种无色透明树脂薄膜的制造方法,其为通过溶液流延法而制造树脂薄膜的方法,所述溶液流延法包括使树脂的有机溶剂溶液在支撑体上流延并干燥的工序,前述有机溶剂含有沸点80℃以下的有机溶剂(s1)和沸点130℃以上的有机溶剂(s2)各1种以上。
20.发明的效果
21.根据本发明的方法,可以制造透明性和光学各向同性优异的无色透明树脂薄膜。以本发明的方法得到的无色透明树脂薄膜的总透光率、黄色指数(yi)值、雾度等光学特性良好,且表示光学各向同性的延迟量低。因此,以本发明的方法得到的无色透明树脂薄膜作为液晶显示元件、有机el显示元件的透明基板、触摸面板的透明导电薄膜的基材使用的情况下,显示良好的性能。
具体实施方式
22.本发明的无色透明树脂薄膜的制造方法为一种无色透明树脂薄膜的制造方法,其为通过溶液流延法而制造树脂薄膜的方法,所述溶液流延法包括使树脂的有机溶剂溶液在支撑体上流延并干燥的工序,前述有机溶剂含有沸点80℃以下的有机溶剂(s1)和沸点130℃以上的有机溶剂(s2)各1种以上。
23.本发明中的“无色透明”是指,形成厚度20~50μm的薄膜时,薄膜的总透光率优选85%以上、且黄色指数(yi)优选5以下、雾度优选2%以下。
24.作为本实施方式中能使用的树脂,只要可以通过包括使树脂的有机溶剂溶液在支撑体上流延并干燥的工序的溶液流延法而制造树脂薄膜就没有特别限定,树脂薄膜的树脂优选聚酰亚胺。树脂的有机溶剂溶液优选聚酰胺酸的有机溶剂溶液或聚酰亚胺的有机溶剂溶液。以下,以树脂薄膜的树脂为聚酰亚胺的情况为代表例对本实施方式详细地进行说明。
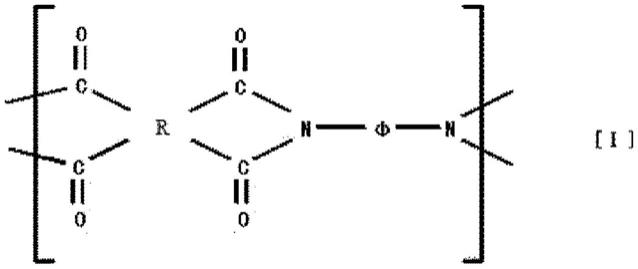
25.作为本实施方式中能使用的树脂薄膜的树脂,从透明性、光学各向同性的方面出发,例如更优选包含下述式[i]所示的重复单元的聚酰亚胺。
[0026][0027]
(式中,r为碳数4~39的4价的脂环基,φ为总计的碳数为2~39的2价的脂肪族基
团、脂环基、芳香族基团或由它们的组合构成的基团,且任选具有选自由
‑
o
‑
、
‑
so2‑
、
‑
co
‑
、
‑
ch2‑
、
‑
c(ch3)2‑
、
‑
osi(ch3)2‑
、
‑
c2h4o
‑
和
‑
s
‑
组成的组中的至少1者作为键合基团。)
[0028]
聚酰亚胺中的式[i]的重复单元的含量相对于聚酰亚胺的全部重复单元100摩尔%,优选10~100摩尔%、更优选50~100摩尔%、进一步优选70~100摩尔%、更进一步优选90~100摩尔%。另外,聚酰亚胺1分子中的式[i]的重复单元的个数优选10~2000、更优选20~200。
[0029]
聚酰亚胺将4价的脂环式四羧酸和2价的二胺作为构成成分,通过使脂环式四羧酸或其衍生物与二胺或其衍生物反应而得到。作为脂环式四羧酸或其衍生物,可以举出脂环式四羧酸、脂环式四羧酸酯类、脂环式四羧酸二酐等,优选脂环式四羧酸二酐。作为二胺和其衍生物,可以举出二胺、二异氰酸酯、二氨基二硅烷类等,优选二胺。
[0030]
作为聚酰亚胺的合成中使用的脂环式四羧酸二酐,可以示例1,2,3,4
‑
环丁烷四羧酸二酐、1,2,4,5
‑
环戊烷四羧酸二酐、1,2,4,5
‑
环己烷四羧酸二酐、双环[2,2,2]辛
‑7‑
烯
‑
2,3,5,6
‑
四羧酸二酐等,特别优选的是,1,2,4,5
‑
环己烷四羧酸二酐。通常,对于将脂肪族二胺作为构成成分的聚酰亚胺,由于作为中间产物的聚酰胺酸与二胺形成牢固的络合物而不易高分子化,因此,需要在使用络合物的溶解性较高的溶剂(例如甲酚)等方面下工夫。然而,将1,2,4,5
‑
环己烷四羧酸二酐和脂肪族二胺作为构成成分的聚酰亚胺中,聚酰胺酸与二胺的络合物以较弱的键连接,因此,高分子量化容易,容易得到柔性的薄膜。需要说明的是,前述四羧酸成分包含异构体。
[0031]
上述四羧酸成分中,在不有损聚酰亚胺的溶剂可溶性、薄膜的挠性、透明性的范围内,可以组合使用除脂环式四羧酸以外的四羧酸或其衍生物、特别是二酐。
[0032]
作为除脂环式四羧酸以外的四羧酸,可以举出芳香族四羧酸和直链或支链的脂肪族四羧酸。作为芳香族四羧酸的具体例,可以举出选自均苯四酸、3,3’,4,4
’‑
联苯四羧酸、2,3,3’,4
’‑
联苯四羧酸、2,2
‑
双(3,4
‑
二羧基苯基)丙烷、2,2
‑
双(2,3
‑
二羧基苯基)丙烷、2,2
‑
双(3,4
‑
二羧基苯基)
‑
1,1,1,3,3,3
‑
六氟丙烷、2,2
‑
双(2,3
‑
二羧基苯基)
‑
1,1,1,3,3,3
‑
六氟丙烷、双(3,4
‑
二羧基苯基)砜、双(3,4
‑
二羧基苯基)醚、双(2,3
‑
二羧基苯基)醚、3,3’,4,4
’‑
二苯甲酮四羧酸、2,2’,3,3
’‑
二苯甲酮四羧酸、4,4
‑
(对亚苯基二氧基)二苯二甲酸、4,4
‑
(间亚苯基二氧基)二苯二甲酸、1,1
‑
双(2,3
‑
二羧基苯基)乙烷、双(2,3
‑
二羧基苯基)甲烷、双(3,4
‑
二羧基苯基)甲烷和这些四羧酸的衍生物、特别是二酐中的至少1种化合物。作为直链或支链的脂肪族四羧酸的具体例,可以举出亚乙基四羧酸等。
[0033]
作为构成式[i]的酰亚胺环的氮和φ的二胺系成分,可以举出二胺、二异氰酸酯、二氨基二硅烷类等,优选二胺。二胺系成分中的二胺含量优选50摩尔%以上、更优选70摩尔%以上、进一步优选90摩尔%以上(包含100摩尔%)。
[0034]
聚酰亚胺的合成中使用的二胺可以为芳香族二胺、脂肪族二胺或它们的混合物,均可。需要说明的是,本发明中“芳香族二胺”是指,氨基直接键合于芳香族环的二胺,在其结构的一部分中可以包含脂肪族基团、脂环基、其他取代基。“脂肪族二胺”是指,氨基直接键合于脂肪族基团或脂环基的二胺,在其结构的一部分中可以包含芳香族基团、其他取代基。
[0035]
作为聚酰亚胺的合成中使用的芳香族二胺,例如可以举出:对苯二胺、间苯二胺、2,4
‑
二氨基甲苯、2,6
‑
二氨基甲苯、联苯胺、邻联甲苯胺、间联甲苯胺、双(三氟甲基)联苯
胺、八氟联苯胺、3,3
’‑
二羟基
‑
4,4
’‑
二氨基联苯、3,3
’‑
二甲氧基
‑
4,4
’‑
二氨基联苯、3,3
’‑
二氯
‑
4,4
’‑
二氨基联苯、3,3
’‑
二氟
‑
4,4
’‑
二氨基联苯、2,6
‑
二氨基萘、1,5
‑
二氨基萘、4,4
’‑
二氨基二苯醚、3,4
’‑
二氨基二苯醚、4,4
’‑
二氨基二苯基甲烷、4,4
’‑
二氨基二苯基砜、3,4
’‑
二氨基二苯基砜、4,4
’‑
二氨基二苯甲酮、2,2
‑
双(4
‑
(4
‑
氨基苯氧基)苯基)丙烷、2,2
‑
双(4
‑
(2
‑
甲基
‑4‑
氨基苯氧基)苯基)丙烷、2,2
‑
双(4
‑
(2,6
‑
二甲基
‑4‑
氨基苯氧基)苯基)丙烷、2,2
‑
双(4
‑
(4
‑
氨基苯氧基)苯基)六氟丙烷、2,2
‑
双(4
‑
(2
‑
甲基
‑4‑
氨基苯氧基)苯基)六氟丙烷、2,2
‑
双(4
‑
(2,6
‑
二甲基
‑4‑
氨基苯氧基)苯基)六氟丙烷、4,4
’‑
双(4
‑
氨基苯氧基)联苯、4,4
’‑
双(2
‑
甲基
‑4‑
氨基苯氧基)联苯、4,4
’‑
双(2,6
‑
二甲基
‑4‑
氨基苯氧基)联苯、4,4
’‑
双(3
‑
氨基苯氧基)联苯、双(4
‑
(4
‑
氨基苯氧基)苯基)砜、双(4
‑
(2
‑
甲基
‑4‑
氨基苯氧基)苯基)砜、双(4
‑
(2,6
‑
二甲基
‑4‑
氨基苯氧基)苯基)砜、双(4
‑
(4
‑
氨基苯氧基)苯基)醚、双(4
‑
(2
‑
甲基
‑4‑
氨基苯氧基)苯基)醚、双(4
‑
(2,6
‑
二甲基
‑4‑
氨基苯氧基)苯基)醚、1,4
‑
双(4
‑
氨基苯氧基)苯、1,4
‑
双(2
‑
甲基
‑4‑
氨基苯氧基)苯、1,4
‑
双(2,6
‑
二甲基
‑4‑
氨基苯氧基)苯、1,3
‑
双(4
‑
氨基苯氧基)苯、1,3
‑
双(2
‑
甲基
‑4‑
氨基苯氧基)苯、1,3
‑
双(2,6
‑
二甲基
‑4‑
氨基苯氧基)苯、2,2
‑
双(4
‑
氨基苯基)丙烷、2,2
‑
双(2
‑
甲基
‑4‑
氨基苯基)丙烷、2,2
‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)丙烷、2,2
‑
双(4
‑
氨基苯基)六氟丙烷、2,2
‑
双(2
‑
甲基
‑4‑
氨基苯基)六氟丙烷、2,2
‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)六氟丙烷、α,α
’‑
双(4
‑
氨基苯基)
‑
1,4
‑
二异丙基苯、α,α
’‑
双(2
‑
甲基
‑4‑
氨基苯基)
‑
1,4
‑
二异丙基苯、α,α
’‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)
‑
1,4
‑
二异丙基苯、α,α
’‑
双(3
‑
氨基苯基)
‑
1,4
‑
二异丙基苯、α,α
’‑
双(4
‑
氨基苯基)
‑
1,3
‑
二异丙基苯、α,α
’‑
双(2
‑
甲基
‑4‑
氨基苯基)
‑
1,3
‑
二异丙基苯、α,α
’‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)
‑
1,3
‑
二异丙基苯、α,α
’‑
双(3
‑
氨基苯基)
‑
1,3
‑
二异丙基苯、9,9
‑
双(4
‑
氨基苯基)芴、9,9
‑
双(2
‑
甲基
‑4‑
氨基苯基)芴、9,9
‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)芴、1,1
‑
双(4
‑
氨基苯基)环戊烷、1,1
‑
双(2
‑
甲基
‑4‑
氨基苯基)环戊烷、1,1
‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)环戊烷、1,1
‑
双(4
‑
氨基苯基)环己烷、1,1
‑
双(2
‑
甲基
‑4‑
氨基苯基)环己烷、1,1
‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)环己烷、1,1
‑
双(4
‑
氨基苯基)4
‑
甲基
‑
环己烷、1,1
‑
双(4
‑
氨基苯基)降冰片烷、1,1
‑
双(2
‑
甲基
‑4‑
氨基苯基)降冰片烷、1,1
‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)降冰片烷、1,1
‑
双(4
‑
氨基苯基)金刚烷、1,1
‑
双(2
‑
甲基
‑4‑
氨基苯基)金刚烷、1,1
‑
双(2,6
‑
二甲基
‑4‑
氨基苯基)金刚烷等。
[0036]
进而,作为聚酰亚胺的合成中使用的脂肪族二胺,例如可以举出:乙二胺、六亚甲基二胺、聚乙二醇双(3
‑
氨基丙基)醚、聚丙二醇双(3
‑
氨基丙基)醚、1,3
‑
双(氨基甲基)环己烷、1,4
‑
双(氨基甲基)环己烷、间苯二甲胺、对苯二甲胺、1,4
‑
双(2
‑
氨基
‑
异丙基)苯、1,3
‑
双(2
‑
氨基
‑
异丙基)苯、异佛尔酮二胺、降冰片烷二胺、硅氧烷二胺类等。
[0037]
聚酰亚胺通常作为有机溶剂溶液(清漆)而制造。本发明的方法中,作为有机溶剂,使用含有沸点80℃以下的有机溶剂(s1)和沸点130℃以上的有机溶剂(s2)各1种以上的有机溶剂。本发明中,通过组合使用具有较低的沸点的有机溶剂(s1)和具有较高的沸点的有机溶剂(s2),从而在干燥工序中,使有机溶剂(s1)有效地挥发,抑制薄膜的黄变,且敢于使有机溶剂(s2)残留于薄膜中从而可以得到光学各向同性优异的薄膜。
[0038]
有机溶剂(s1)的沸点为80℃以下、优选75℃以下、更优选70℃以下、进一步优选65℃以下、进一步优选60℃以下。有机溶剂(s1)的沸点的下限值没有特别限定,从作业效率的观点出发,优选30℃以上。通过使用具有80℃以下的较低的沸点的有机溶剂(s1),从而在
120~150℃左右的较低的干燥温度下使有机溶剂(s1)挥发,可以得到树脂薄膜。
[0039]
作为有机溶剂(s1),没有特别限定,例如可以使用二氯甲烷(dcm、沸点39.6℃)、1,3
‑
二氧戊环(沸点75℃)、四氢呋喃(thf、沸点66℃)、丙酮(沸点56℃)、氯仿(沸点61℃)、乙酸乙酯(沸点77℃)等,可以将2种以上组合使用。它们之中,作为有机溶剂(s1),从聚酰亚胺清漆的性能的观点出发,优选选自由二氯甲烷和1,3
‑
二氧戊环组成的组中的至少1种。
[0040]
有机溶剂(s2)的沸点为130℃以上、优选140℃以上、更优选150℃以上、进一步优选160℃以上。有机溶剂(s2)的沸点的上限值没有特别限定,例如可以为300℃以下,也可以为250℃以下。通过使用具有130℃以上的较高的沸点的有机溶剂(s2),从而在120~150℃左右的干燥温度下使树脂的有机溶剂溶液干燥时,可以得到延迟量低、光学各向同性优异的树脂薄膜。其理由尚不清楚,但推定:通过在120~150℃左右的较低的干燥温度下使树脂的有机溶剂溶液干燥,从而在树脂薄膜中残留有使用上没有问题的程度的有机溶剂(s2),由此,高分子链的取向得到缓和,可以实现低的延迟量。
[0041]
作为有机溶剂(s2),没有特别限定,例如可以使用环戊酮(沸点131℃)、环己酮(沸点156℃)、n
‑
甲基
‑2‑
吡咯烷酮(沸点202℃)、n,n
‑
二甲基乙酰胺(dmac、沸点165℃)、n,n
‑
二甲基甲酰胺(沸点153℃)、γ
‑
丁内酯(gbl、沸点204℃)、二甲基亚砜(沸点189℃)、二甲基异丁酰胺(沸点179℃)等,可以将2种以上组合使用。它们之中,作为有机溶剂(s2),从聚酰亚胺清漆的性能的观点出发,优选选自由n
‑
甲基
‑2‑
吡咯烷酮、n,n
‑
二甲基乙酰胺、γ
‑
丁内酯和二甲基亚砜组成的组中的至少1种。
[0042]
从在较低的干燥温度下使树脂的有机溶剂溶液干燥、抑制薄膜的黄变的观点出发,作为聚酰亚胺的有机溶剂溶液中所含的有机溶剂(s1)与有机溶剂(s2)的质量比[(s1)/(s2)]优选90/10~99/1、更优选93/7~97/3。
[0043]
作为聚酰亚胺的有机溶剂溶液的制造方法,可以举出下述(1)~(3)的方法,但不限定于这些方法。
[0044]
(1)在二胺系成分的有机溶剂溶液中添加四羧酸成分,或在四羧酸成分的有机溶剂溶液中添加二胺系成分,在优选80℃以下、特别是在室温附近或其以下的温度下保持0.5~3小时。在得到的反应中间体的聚酰胺酸溶液中添加甲苯或二甲苯等共沸脱水溶剂,将生成的水通过共沸去除至体系外,且进行脱水反应,得到聚酰亚胺的有机溶剂溶液。
[0045]
(2)在与上述(1)同样地得到的反应中间体的聚酰胺酸溶液中加入乙酸酐等脱水剂进行酰亚胺化后,添加甲醇等对聚酰亚胺缺乏溶解能力的溶剂,使聚酰亚胺沉淀。通过过滤、清洗和干燥而作为固体分离后,溶解于有机溶剂,得到聚酰亚胺的有机溶剂溶液。
[0046]
(3)上述(1)中,使用甲酚等高沸点溶剂制备聚酰胺酸溶液,直接在150~220℃下保持3~12小时,进行聚酰亚胺化后,添加甲醇等对聚酰亚胺缺乏溶解能力的溶剂,使聚酰亚胺沉淀。通过过滤、清洗和干燥而作为固体分离后,溶解于有机溶剂,得到聚酰亚胺的有机溶剂溶液。
[0047]
另外,以溶液聚合制造聚酰亚胺的情况下,优选使用叔胺化合物作为催化剂。作为这些,可以举出三甲胺、三乙胺(tea)、三丙胺、三丁胺、三乙醇胺、n,n
‑
二甲基乙醇胺、n,n
‑
二乙基乙醇胺、三乙二胺、n
‑
甲基吡咯烷、n
‑
乙基吡咯烷、n
‑
甲基哌啶、n
‑
乙基哌啶、咪唑、吡啶、喹啉、异喹啉等。这些叔胺化合物中,特别优选tea。
[0048]
另外,本发明中使用的聚酰亚胺的有机溶剂溶液的浓度如下:聚酰亚胺成分优选4
~45质量%、更优选10~40质量%。具体而言,树脂的有机溶剂溶液含有相对于有机溶剂100g、以优选5~80g、更优选10~70g的范围内溶解的树脂。如果为该范围内,则得到的聚酰亚胺薄膜的表面平滑性良好。
[0049]
从得到的聚酰亚胺的弯曲性、机械强度的观点出发,本发明中使用的聚酰亚胺的重均分子量优选10000以上、更优选50000以上。需要说明的是,聚酰亚胺的重均分子量可以用公知的方法测定,例如可以通过凝胶渗透色谱法等测定。另外,还可以举出展开溶剂中使用n,n
‑
二甲基甲酰胺、用光散射检出器测定绝对分子量的方法。
[0050]
聚酰亚胺的有机溶剂溶液中,可以添加氟系、聚硅氧烷系等表面活性剂。通过添加表面活性剂,从而变得容易得到表面平滑性良好的薄膜。
[0051]
聚酰亚胺的有机溶剂溶液中,可以添加酚系、硫系、磷酸系、亚磷酸系等的抗氧化剂。
[0052]
本发明的聚酰亚胺薄膜的制造方法没有特别限制,可以使用公知的方法。例如可以举出如下方法等:将包含本发明的聚酰亚胺的溶液、或将包含本发明的聚酰亚胺的溶液和包含上述各种添加剂的溶液涂布于玻璃板、金属板、塑料等平滑的支撑体上、或成型为薄膜状后,去除该溶液中所含的有机溶剂等溶剂成分。
[0053]
作为无色透明聚酰亚胺薄膜的制造方法,可以举出如下方法:以使聚酰亚胺的有机溶剂溶液在支撑体上流延并干燥的溶液流延法形成薄膜。具体而言,使聚酰亚胺的有机溶剂溶液在支撑体上流延后,使用向支撑体上的流延物吹送优选80℃以上且250℃以下的气体的形式的制膜机,使有机溶剂挥发,作为自支撑性薄膜可以从支撑体剥离。优选在吹送气体前进行一次干燥。一次干燥的条件没有特别限定,例如优选在80~120℃的温度下保持10~30分钟。
[0054]
作为吹送的气体,可以举出空气或氮气,从成本的观点出发,优选空气,从防止薄膜的着色的观点出发,优选氮气。吹送的气体的温度更优选80℃以上且250℃以下、进一步优选100℃以上且220℃以下。吹送的气体的温度低于80℃的情况下,有机溶剂不充分挥发,将薄膜从支撑体剥离时,有时发生对支撑体的粘附等。另外,气体的温度高于250℃的情况下,溶剂急剧挥发,因此,薄膜中产生发泡,另外,溶剂分解而薄膜有时着色。吹送气体的时间根据吹送的气体的温度而不同,优选15~30分钟、更优选15~25分钟。另外,也可以设置向流延物吹送的气体的温度不同的多个区域。
[0055]
另外,本发明的无色透明聚酰亚胺薄膜的制造方法中,使树脂的有机溶剂溶液在支撑体上流延并干燥的工序优选包括如下工序:工序(i),将前述溶剂的一部分去除使得通过差热/热重同时测定求出的120~300℃下的重量减少率成为超过1%且10%以下;和,工序(ii),将前述溶剂的一部分去除的工序(i)后,将树脂的玻璃化转变温度设为tg(℃)时,在(tg
‑
50)~(tg+100)℃的范围内进行热处理。
[0056]
前述工序(i)中的前述重量减少率优选超过1%且10%以下、更优选1%以上且5%以下、进一步优选1%以上且3%以下。如果为该范围内,则干燥不过度耗费时间,可以制造透明性和光学各向同性优异的无色透明树脂薄膜。
[0057]
前述工序(ii)中的热处理温度优选(tg
‑
50)℃以上且(tg+100)℃以下、更优选(tg
‑
30)℃以上且(tg+80)℃以下、进一步优选(tg
‑
10)℃以上且(tg+60)℃以下。如果为该范围内,则可以制造透明性和光学各向同性优异的无色透明树脂薄膜。
[0058]
聚酰亚胺薄膜在不有损透明性和弯曲性的范围内可以还含有其他成分。作为其他成分,例如可以举出增塑剂、抗氧化剂、脱模剂、稳定剂、蓝化剂等着色剂、阻燃剂、润滑剂、增稠剂、和流平剂等。
[0059]
本制造方法得到的聚酰亚胺薄膜的厚度可以根据用途而适宜调整,通常为10~500μm、优选15~200μm、更优选20~100μm。
[0060]
对于该聚酰亚胺薄膜,在厚度20~50μm下,依据jis k7361
‑
1的总透光率优选85%以上、更优选90%以上。
[0061]
另外,对于该聚酰亚胺薄膜,在厚度20~50μm下,依据jis k7361
‑
1的雾度优选2%以下、更优选1%以下。
[0062]
另外,对于该聚酰亚胺薄膜,在厚度20~50μm下,依据jis k7361
‑
1的黄色指数(yi)优选5以下、更优选3以下。
[0063]
对于该聚酰亚胺薄膜,厚度方向的延迟量(rth)优选50nm以下、更优选40nm以下、进一步优选30nm以下。
[0064]
对于该聚酰亚胺薄膜,面内的延迟量(re)优选20nm以下、更优选15nm以下。
[0065]
以本制造方法得到的聚酰亚胺薄膜可以包含添加剂等作为其他成分。例如通过包含二氧化钛等,从而改善白色光的反射率。另外,通过包含纳米填料等,从而树脂组成物成型体的表观玻璃化转变温度上升,耐热性提高,进一步拉伸模量变大,机械强度增大。
[0066]
以本发明的方法得到的无色透明树脂薄膜适合用作触摸传感器、滤色器、柔性显示器、半导体部件、光学构件等各种构件用的薄膜。另外,以本发明的方法得到的无色透明树脂薄膜作为液晶显示元件、有机el显示元件的透明基板、触摸面板的透明导电薄膜的基材是有用的。
[0067]
实施例
[0068]
以下,根据实施例,对本发明具体地进行说明。但本发明不受这些实施例的任何限制。
[0069]
以下示出下述实施例中得到的薄膜的物性的测定方法。
[0070]
(1)薄膜厚度
[0071]
薄膜厚度用mitutoyo co.,ltd.制的测微器而测定。
[0072]
(2)总透光率、雾度、黄色指数(yi)
[0073]
测定依据jis k7361
‑
1,用日本电色工业株式会社制的色彩/浊度同时测定器“coh400”而进行。
[0074]
(3)面内延迟量(re)
[0075]
面内延迟量(re)用日本分光株式会社制的椭偏仪“m
‑
220”而测定。测定在测定波长590nm下的面内相位差的值。
[0076]
(4)厚度方向延迟量(rth)
[0077]
厚度相位差(rth)用日本分光株式会社制的椭偏仪“m
‑
220”而测定。测定在测定波长590nm下的厚度相位差的值。需要说明的是,将聚酰亚胺薄膜的面内的折射率中的最大者设为nx、最小者设为ny、厚度方向的折射率设为nz、薄膜的厚度设为d时,rth由下述式表示。
[0078]
rth=[{(nx+ny)/2}
‑
nz]
×
d
[0079]
(5)薄膜中的有机溶剂含量
[0080]
用hitachi high
‑
tech science corporation制的差热/热重同时测定装置“tg/dta6200”,在氮气气流下、在升温速度10℃/分钟的条件下进行测定,从120℃升温至300℃,然后在300℃下保持30分钟,将在此期间减少的质量作为薄膜中的有机溶剂含量。
[0081]
(6)玻璃化转变温度(tg)
[0082]
用hitachi high
‑
tech science corporation制的热机械分析装置“tma/ss6100”,在拉伸模式下,在试样尺寸2mm
×
20mm、载荷0.1n、升温速度10℃/分钟的条件,升温至充分去除残留应力的温度,去除残留应力,之后冷却至室温。之后,在与用于去除前述残留应力的处理相同的条件下进行试验片伸长率的测定,将可见伸长率的拐点之处作为玻璃化转变温度而求出。
[0083]
<实施例1>
[0084]
在具备不锈钢制半月型搅拌叶片、氮气导入管、装有冷凝管的迪安
‑
斯达克榻分水器、温度计、玻璃制端盖的2l的五口玻璃制圆底烧瓶中,将α,α
’‑
双(4
‑
氨基苯基)
‑
1,3
‑
二异丙基苯(mitsui chemical fine co.,ltd.制)239.772g(0.696摩尔)、4,4
‑
二氨基二苯醚(和歌山精化工业株式会社制)34.842g(0.174摩尔)、γ
‑
丁内酯(三菱化学株式会社制)376.453g、和作为催化剂的三乙胺(关东化学株式会社制)44.018g、三乙二胺(东京化成工业株式会社制)0.488g在反应体系内温度70℃、氮气气氛下、以200rpm搅拌,得到溶液。向其中分别一次性加入1,2,4,5
‑
环己烷四羧酸二酐(三菱瓦斯化学株式会社制)195.028g(0.870摩尔)和γ
‑
丁内酯(三菱化学株式会社制)94.113g后,在有罩加热器中加热,用约20分钟,使反应体系内温度升高至200℃。捕集蒸馏去除的成分,根据粘度上升调整搅拌数,且将反应体系内温度维持在200℃下5小时。添加n,n
‑
二甲基乙酰胺847.067g后,在100℃附近搅拌约1小时,形成均匀的溶液,得到固体成分浓度25质量%的均匀的聚酰亚胺清漆(a)。
[0085]
接着,在甲醇中滴加得到的聚酰亚胺清漆,使聚酰亚胺粉末沉淀,将固体用桐山漏斗抽滤,进一步用甲醇清洗,以200℃进行30分钟干燥处理从而去除溶剂,得到聚酰亚胺粉末。
[0086]
在具备不锈钢制半月型搅拌叶片、氮气导入管、装有冷凝管的迪安
‑
斯达克榻分水器、温度计、玻璃制端盖的300ml的五口玻璃制圆底烧瓶中,一次性加入得到的聚酰亚胺粉末15g、二氯甲烷(dcm)80.75g和n,n
‑
二甲基乙酰胺(dmac)4.25g后,在室温下搅拌1小时,形成均匀的溶液,得到固体成分浓度15质量%的均匀的聚酰亚胺清漆(b)。
[0087]
接着,将得到的聚酰亚胺清漆(b)涂布于pet基板上,在室温下保持5分钟后,以50℃、在空气气氛下保持5分钟,最后在空气气氛下吹送150℃的热风30分钟并干燥,从而得到厚度35μm的薄膜。将该聚酰亚胺薄膜的评价结果示于表1。
[0088]
<实施例2>
[0089]
对实施例1中得到的聚酰亚胺薄膜在空气气氛下吹送250℃的热风20分钟并进一步干燥,从而得到厚度35μm的薄膜。将该聚酰亚胺薄膜的评价结果示于表1。
[0090]
<比较例1>
[0091]
在甲醇中滴加实施例1中得到的聚酰亚胺清漆(a),使聚酰亚胺粉末沉淀,将固体用桐山漏斗抽滤,进一步用甲醇清洗,以200℃进行30分钟干燥处理,从而去除溶剂,得到聚酰亚胺粉末。
[0092]
在具备不锈钢制半月型搅拌叶片、氮气导入管、装有冷凝管的迪安
‑
斯达克榻分水
器、温度计、玻璃制端盖的300ml的五口玻璃制圆底烧瓶中,一次性加入得到的聚酰亚胺粉末15g和二氯甲烷(dcm)85g后,在室温下搅拌1小时形成均匀的溶液,得到固体成分浓度15质量%的均匀的聚酰亚胺清漆。
[0093]
接着,将得到的聚酰亚胺清漆涂布于pet基板上,在室温下保持5分钟后,以50℃、在空气气氛下保持5分钟,最后在空气气氛下吹送150℃的热风20分钟并干燥,从而得到厚度35μm的薄膜。将该聚酰亚胺薄膜的评价结果示于表1。
[0094]
<比较例2>
[0095]
对比较例1中得到的聚酰亚胺薄膜在空气气氛下吹送250℃的热风20分钟并进一步干燥,从而得到厚度35μm的薄膜。将该聚酰亚胺薄膜的评价结果示于表1。
[0096]
[表1]
[0097]
表1
[0098][0099]
如表1所示,实施例1和2中得到的聚酰亚胺薄膜的总透光率、雾度、yi等光学特性良好,且rth低,可以得到光学各向同性优异的薄膜。与此相对,比较例1和2中得到的聚酰亚胺薄膜的rth大、光学各向同性差。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1