用于修饰蛋白质和/或肽的分子
1.本发明涉及用于修饰蛋白质和/或肽等的分子。
背景技术:
2.将蛋白质和/或肽(以下简称“蛋白质等”)与其他分子/物质连接的技术在制备抗体
‑
药物偶联物、荧光探针标记的蛋白质试剂、蛋白质固定无机材料等中很重要。例如,用叠氮基修饰蛋白质等是一种能够通过炔
‑
叠氮环加成反应(cuaac)引入各种功能分子的技术。由于生物正交性,该技术已广泛应用于生物成像等领域。
3.引文列表
4.非专利文献
5.非专利文献1:metal
‑
free and ph
‑
controlled introduction of azides in proteins,sanne schoffelen,mark b.van eldijk,bart rooijakkers,reinout raijmakers,albert j.r.heck and jan c.m.van hest,chemical science,2011,2,701.
6.非专利文献2:selective n
‑
terminal acylation of peptides andproteins with agly
‑
his tag sequence,m.c.martos
‑
maldonado,c.t.hjuler,k.k.sorensen,m.b.thygesen,j.e.rasmussen,k.villadsen,s.r.midtgaard,s.kol,s.schoffelen,k.j.jensen,nature communications,2018,9,3307.
7.非专利文献3:modification of n
‑
terminalα
‑
amino groups of peptides and proteins using ketenes,a.o.
‑
y.chan,c.
‑
m.ho,h.
‑
c.chong,y.
‑
c.leung,j.
‑
s.huang,m.
‑
k.wong,c.
‑
m.che,journal of the american chemical society,2012,134,2589.
技术实现要素:
8.技术问题
9.本发明人在研究将其他分子/物质与蛋白质等连接时的引入位置时,着眼于以下三点。第一点是所有单体蛋白质都只有一个n端位置,这是一个通用的修饰基点。第二点是n端很少参与蛋白质活性位点(分子结合位点和催化反应中心)或蛋白质功能位点,与修饰相关的结构变化的影响被认为很小。第三点是,由于pk
a
的差异,n端被认为不太可能与其他氨基酸残基(例如赖氨酸、半胱氨酸和谷氨酰胺)竞争反应;而在c端,这种基于pk
a
的反应选择性被认为是难以实现的。因此,发明人集中于蛋白质等的n端作为连接其他分子/物质的位置。
10.将其他分子/物质连接到蛋白质等的n端的各种方法已经被报道过了(非专利文献1至非专利文献3)。然而,这些方法在简单性或n端修饰选择性方面被认为是不够的。例如,虽然识别特定氨基酸序列的化学键合方法或脂质修饰酶方法可以将其他分子/物质特异性引入n端,但它们需要其中插入特定氨基酸序列或特定氨基酸残基的蛋白质等。这样的蛋白质等的制备是费力的,并且这些方法不能应用于天然蛋白质等。使用活化酯或乙烯酮的酰胺键形成反应操作简单,可应用于天然蛋白质等;然而,它不能将其他分子/物质特异性地
引入到n端,因为会发生与赖氨酸残基等的副反应。
11.因此,本发明的一个目的是提供一种技术,该技术允许其他分子/物质以简单有效的方式更有选择性地连接到所有蛋白质和/或肽的n端。
12.问题解决方案
13.本发明人为解决上述问题进行了广泛的研究。他们发现可以通过使式(1)表示的化合物或其盐,或者该化合物或其盐的水合物或溶剂合物与蛋白质和/或肽反应来解决这些问题。本发明人基于该发现进行了进一步的研究,从而完成了本发明。
14.具体地,本发明包括以下实施方案。
15.第1项.由式(1)表示的化合物或其盐,或该化合物或其盐的水合物或溶剂合物,
[0016][0017]
其中r1和r2之一表示
‑
n(
‑
r4)
‑
(其中r4表示有机基团或衍生自无机材料的基团),另一个表示=n
‑
;且r3表示氢原子、有机基团或衍生自无机材料的基团。
[0018]
第2项.根据第1项所述的化合物或其盐,或者所述化合物或其盐的水合物或溶剂合物,其中所述化合物由式(1aa)表示:
[0019][0020]
其中r4如上所定义。
[0021]
第3项.根据第1项或第2项所述的化合物或其盐,或该化合物或其盐的水合物或溶剂合物,其中所述有机基团为衍生自有机分子或有机分子复合物的基团,并且所述有机分子或有机分子复合物是功能性物质。
[0022]
第4项.根据第3项所述的化合物或其盐,或所述化合物或其盐的水合物或溶剂合物,其中所述功能性物质为药物化合物、发光分子、高分子化合物、配体、配体结合的分子、抗原性蛋白质、抗体、蛋白质、核酸、糖类、脂质、细胞、病毒、标签、碳电极、碳纳米材料、连接体、间隔分子,或其复合物或连接分子。
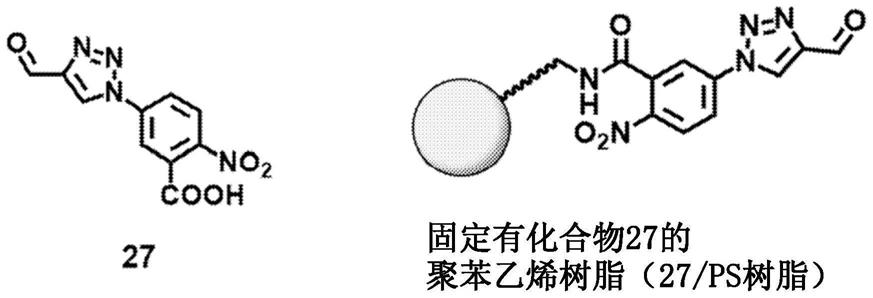
[0023]
第5项.根据第1至4项中任一项所述的化合物或其盐,或所述化合物或其盐的水合物或溶剂合物,其中所述无机材料为电极材料、金属微粒、金属氧化物微粒、半导体粒子或磁性粒子。
[0024]
第6项.一种试剂,其包含第1至5项中任一项所述的化合物或其盐,或所述化合物或其盐的水合物或溶剂合物。
[0025]
第7项.根据第6项所述的试剂,其是用于蛋白质和/或肽修饰的试剂。
[0026]
第8项.一种制备根据第1至5项中任一项所述的化合物或其盐、或所述化合物或其盐的水合物或溶剂合物的方法,该方法包括:
[0027]
使式(2)表示的化合物或其盐与式(3)表示的化合物、式(4)表示的化合物或式(9)表示的化合物反应,
[0028]
r4‑
r5(2),
[0029]
其中r4表示有机基团或衍生自无机材料的基团,且r5表示
‑
n3、
‑
x(其中x表示卤素原子),
‑
b(oh)2,
‑
b(or
51
)2(其中每个r
51
相同或不同并表示一个烃基,条件是两个r
51
与相邻的氧原子一起可以形成一个环),或
‑
n
2+
,
[0030][0031]
其中r3表示氢原子、有机基团或衍生自无机材料的基团;r6、r7和r8相同或不同,且各自表示烷基;并且y表示反应性基团。
[0032]
第9项.一种制备根据第1至5项中任一项所述的化合物或其盐、或所述化合物或其盐的水合物或溶剂合物的方法,该方法包括:
[0033]
使式(5)表示的化合物与式(6)表示的化合物反应:
[0034]
r4‑
r9(5),
[0035]
其中r4表示有机基团或衍生自无机材料的基团;且r9表示
‑
nr
9a
r
9b
(其中r
9a
和r
9b
相同或不同且各自表示氢原子或烷基),
[0036][0037]
其中r
10
表示吸电子基团;r
11
表示
‑
r
11a
‑
r
12
(其中r
11a
表示单键或连接体;且r
12
表示载体);n表示0或1;且m表示1~5的整数。
[0038]
第10项.由式(6’)表示的化合物:
[0039][0040]
其中r
10
表示吸电子基团;r
11a
表示单键或连接体;r
12
表示载体;且m表示1~5的整数。
[0041]
第11项.由式(7)表示的化合物或其盐,或该化合物或其盐的水合物或溶剂合物,
[0042][0043]
其中r1和r2之一表示
‑
n(
‑
r4)
‑
(其中r4表示有机基团或衍生自无机材料的基团),另一个表示=n
‑
;r3表示氢原子、有机基团或衍生自无机材料的基团;由虚线和实线组成的双线表示单键或双键;r
13
表示n端氨基酸残基和与其相邻的
‑
nh
‑
被排除在蛋白质或肽之外的基团;r
14
表示蛋白质或肽的n端氨基酸残基的侧链。
[0044]
第12项.一种制备根据第11项的化合物或其盐、或该化合物或其盐的水合物或溶剂合物的方法,该方法包括使蛋白质和/或肽与根据第1至5项中任一项所述的化合物或其盐,或所述化合物或其盐的水合物或溶剂合物反应。
[0045]
发明的有益效果
[0046]
本发明可以提供一种技术,该技术允许其他分子/物质以简单和有效的方式更有选择性地连接到n端,甚至在天然蛋白质等中。
附图说明
[0047]
图1显示了化合物1的1h nmr谱(400mhz,dmso
‑
d6)。
[0048]
图2显示了化合物2的1h nmr谱(400mhz,dmso
‑
d6)。
[0049]
图3显示了化合物3的1h nmr谱(400mhz,dmso
‑
d6)。
[0050]
图4显示了化合物4的1h nmr谱(400mhz,cdcl3)。
[0051]
图5显示了化合物5的1h nmr谱(400mhz,dmso
‑
d6)。
[0052]
图6显示了化合物6的1h nmr谱(400mhz,dmso
‑
d6)。
[0053]
图7显示了化合物7的1h nmr谱(400mhz,dmso
‑
d6)。
[0054]
图8显示了化合物8的1h nmr谱(400mhz,cdcl3)。
[0055]
图9显示了化合物9的1h nmr谱(400mhz,dmso
‑
d6)。
[0056]
图10显示了化合物10的1h nmr谱(400mhz,dmso
‑
d6)。
[0057]
图11显示了化合物13的1h nmr谱(400mhz,dmso
‑
d6)。
[0058]
图12显示了化合物16的1h nmr谱(400mhz,dmso
‑
d6)。
[0059]
图13显示了化合物17的1h nmr谱(400mhz,cd3cn)。
[0060]
图14显示了化合物18的1h nmr谱(400mhz,cd3cn)。
[0061]
图15显示了化合物19的1h nmr谱(400mhz,cd3cn)。
[0062]
图16显示了化合物20的1h nmr谱(400mhz,cdcl3)。
[0063]
图17显示了化合物21的1h nmr谱(400mhz,cdcl3)。
[0064]
图18显示了化合物22的1h nmr谱(400mhz,cd3cn)。
[0065]
图19显示了血管紧张素i的n端修饰反应的反应方案和结果(实施例8)。
[0066]
图20显示了化合物25的1h nmr谱(400mhz,cd3cn)。
[0067]
图21显示了核糖核酸酶a的n端修饰反应的反应方案和结果(实施例10)。
[0068]
图22显示了用生物素对核糖核酸酶a进行修饰的反应方案和结果(实施例11
‑
2)。
[0069]
图23显示了用荧光染料修饰核糖核酸酶a的反应方案和结果(实施例11
‑
3)。
[0070]
图24显示了用叠氮基修饰核糖核酸酶a的反应方案和结果(实施例11
‑
4)。
[0071]
图25显示了通过应变促进的炔
‑
叠氮化物环加成反应用荧光染料修饰的反应方案和结果(实施例11
‑
5)。
[0072]
图26显示了用应变炔部分修饰核糖核酸酶a的反应方案和结果(实施例11
‑
6)。
[0073]
图27显示通过将各种甲醛前体固定在树脂或固体材料上,简化去除反应后生成的副产物(苯胺衍生物),反应后的溶液直接用于蛋白质的n端修饰的方法的总结(实施例12)。
[0074]
图28显示化合物27和固定有化合物27的树脂(实施例12)。
[0075]
图29显示了27/ps树脂的合成方案(实施例12
‑
2)。
[0076]
图30显示了使用红外光谱的树脂鉴定结果(实施例12
‑
3)。右边是放大图。
[0077]
图31显示使用固定有反应物的树脂(27/ps树脂)(实施例12
‑
4)通过连续反应对n端修饰的核糖核酸酶a进行lc/ms分析的结果。
[0078]
图32显示了27/ps树脂的合成方案(实施例12
‑
2)。
[0079]
图33显示了化合物28的1h nmr谱(400mhz,dmso
‑
d6)。
[0080]
图34显示了化合物30的1h nmr谱(400mhz,dmso
‑
d6)。
[0081]
图35显示了使用固定有反应物的树脂(27/ps树脂)(实施例12
‑
4)通过连续反应对n端修饰的方案(实施例12
‑
6)。
[0082]
图36显示了使用固定有反应物的树脂(27/ps树脂)通过连续反应对n端修饰的核糖核酸酶a进行lc/ms分析结果(实施例12
‑
6)。对应修饰蛋白的峰显示为
“●”
(实心圆),对应未修饰蛋白的峰显示为
“○”
(空心圆)。
[0083]
图37显示了在均相体系中使用迪姆罗特重排反应制备三唑甲醛并应用于蛋白质
修饰的方案(实施例13)。
[0084]
图38显示了迪姆罗特重排反应的反应机理(实施例13
‑
2)。
[0085]
图39显示了使用化合物10的迪姆罗特重排反应中酸催化剂的结构(实施例13
‑
2)。
[0086]
图40显示了通过迪姆罗特重排反应制备n端修饰剂,以及连续蛋白质修饰反应的lc
‑
ms分析结果(实施例13
‑
3)。对应修饰蛋白的峰显示为
“●”
(实心圆),对应未修饰蛋白的峰显示为
“○”
(空心圆)。
[0087]
图41为bis
‑
ta4c的制备方案和蛋白质修饰反应(实施例14
‑
2)。
[0088]
图42显示使用二胺作为前体(实施例14
‑
2)用bis
‑
ta4c对核糖核酸酶a的n端修饰进行lc
‑
ms分析的结果。显示了作为前体的二胺的结构和由lc
‑
ms计算的修饰百分比。
[0089]
图43显示了通过肟形成将功能性分子引入蛋白质n端的方案(实施例14
‑
3)。
[0090]
图44显示了用化合物37修饰的核糖核酸酶a的lc
‑
ms分析结果(实施例14
‑
3)。
[0091]
图45表示用化合物38修饰的核糖核酸酶a的sds
‑
page分析结果(实施例14
‑
3)。
[0092]
图46显示了化合物39的1h nmr谱(400mhz,cdcl3)。
[0093]
图47显示了化合物40的1h nmr谱(400mhz,cdcl3)。
[0094]
图48显示了使用化合物40(实施例15
‑
2)的迪姆罗特重排反应的方案。
[0095]
图49显示化合物7在迪姆罗特重排反应(实施例15
‑
2)中的产率。产率由1h nmr测量计算。
[0096]
图50显示了人血清来源的白蛋白的n端修饰反应的反应方案和结果(实施例16
‑
2)。
[0097]
图51显示了人血清来源的白蛋白的半胱氨酸残基修饰的反应方案和结果(实施例17
‑
2)。
[0098]
图52显示了人血清来源的白蛋白的n端修饰的反应方案和结果,其中半胱氨酸残基被修饰(实施例17
‑
3)。
[0099]
图53显示核糖核酸酶_7溶液静置24小时后的lc/ms分析结果(实施例18
‑
2)。
[0100]
图54显示了核糖核酸酶_7中修饰剂释放随时间的变化(实施例18
‑
2)。
[0101]
图55显示了修饰剂释放随核糖核酸酶_7中ph变化的变化(实施例18
‑
3)。
[0102]
图56显示了化合物42的1h nmr谱(400mhz,dmso
‑
d6)。
[0103]
图57显示了化合物43的1h nmr谱(400mhz,cdcl3)。
[0104]
图58显示了化合物44的1h nmr谱(400mhz,cdcl3)。
[0105]
图59显示了化合物45的1h nmr谱(400mhz,cdcl3)。
[0106]
图60显示了化合物46的1h nmr谱(400mhz,cdcl3)。
[0107]
图61显示了化合物47的1h nmr谱(400mhz,cdcl3)。
[0108]
图62显示了化合物48的1h nmr谱(400mhz,cdcl3)。
[0109]
图63显示了化合物49的1h nmr谱(400mhz,cdcl3)。
[0110]
图64显示了化合物50的1h nmr谱(400mhz,cdcl3)。
[0111]
图65显示了化合物51的1h nmr谱(400mhz,cdcl3)。
[0112]
图66显示了血管紧张素i的n端修饰的反应方案和结果(实施例20)。
[0113]
图67显示了核糖核酸酶a的n端修饰的反应方案和结果(实施例21)。
[0114]
图68显示了将乙酰基和叠氮基引入核糖核酸酶a的方案和结果(实施例22
‑
1)。
[0115]
图69显示了通过应变促进的炔
‑
叠氮化物环加成反应用荧光染料进行的修饰和结果(实施例22
‑
2)。
[0116]
图70显示了化合物52的1h nmr谱(400mhz,dmso
‑
d6)。
[0117]
图71显示了化合物53的1h nmr谱(400mhz,cdcl3)。
[0118]
图72显示了用聚乙二醇修饰核糖核酸酶a的方案和结果(实施例23
‑2‑
2)。
[0119]
图73显示了对hsa2进行n端修饰的反应以及用化合物26修饰的反应的方案和结果(实施例23
‑2‑
3)。
具体实施方式
[0120]
在本说明书中,术语“包括”和“包含”包括“包括”、“包含”、“基本上由
……
组成”和“由
……
组成”的概念。
[0121]
1.蛋白质和/或肽修饰分子
[0122]
在一个实施方案中,本发明涉及一种由式(1)表示的化合物或其盐,或该化合物或其盐的水合物或溶剂合物(在本说明书中,这些可以统称为“本发明的修饰分子”):
[0123][0124]
其中r1和r2之一表示
‑
n(
‑
r4)
‑
(其中r4表示有机基团或衍生自无机材料的基团),另一个表示=n
‑
,r3表示氢原子、有机基团或衍生自无机材料的基团。
[0125]
下面描述本发明的修饰分子。
[0126]1‑
1.化合物
[0127]
其中r1和r2之一表示
‑
n(
‑
r4)
‑
(其中r4表示有机基团或衍生自无机材料的基团),另一个表示=n
‑
,由虚线和实线组成的双线表示单键或双键,并且是单键还是双键取决于r1和r2中的哪一个是
‑
n(
‑
r4)
‑
或=n
‑
。
[0128]
具体地,当r1是
‑
n(
‑
r4)
‑
,并且r2是=n
‑
时,式(1)为式(1a):
[0129][0130]
其中r3和r4的定义同上;和
[0131]
当r1为=n
‑
,且r2为
‑
n(
‑
r4)
‑
时,式(1)为式(1b):
[0132][0133]
其中r3和r4的定义同上。
[0134]
在本发明中,优选地,r1是
‑
n(
‑
r4)
‑
并且r2是=n
‑
;即,式(1)是式(1a)。
[0135]
有机基团只要是源自有机分子或有机分子复合体的基团,例如,从有机分子或有机分子复合体中除去一个或多个原子而获得的基团,就没有特别限制。有机分子没有特别限制并且可以是天然的、合成的或人造的。有机分子复合体没有特别限制,并且实例包括其中包括一个或多个有机分子的多个分子连接的复合体(或生物体)。连接的方式没有特别限定,实例包括氢键、静电力、范德华力、疏水键、共价键、配位键等。这些键可以通过连接体形成(具体例子见后面描述的连接体)。有机分子或有机分子复合物优选为功能性物质。其具体实例包括药物化合物、发光分子、大分子化合物、配体、配体结合的分子、抗原性蛋白质、抗体、蛋白质、核酸、糖类、脂质、细胞、病毒、标签(例如放射性同位素标签)、碳电极、碳纳米材料、连接体、间隔分子(例如,聚乙二醇或其衍生物和肽(例如,包含被细胞中的酶切割的氨基酸序列的肽))及其复合物和连接分子。
[0136]
在本发明的一个实施方案中,本发明的一个或多个修饰分子可以作为有机基团中的部分结构连接(例如,在末端)。在这种情况下,本发明的修饰分子的一个例子是式(1aa)表示的化合物:
[0137][0138]
其中r3如上所定义,并且r3在每次出现时相同或不同;r
4a
表示二价有机基团。r
4a
可包含本发明的修饰分子的部分结构。
[0139]
无机材料是包含或不包含一个或多个金属原子的材料,并且没有特别限制。无机材料的实例包括电极材料、金属微粒、金属氧化物微粒、半导体粒子、磁性粒子等。无机材料可以保留有机分子或有机分子复合物。
[0140]
有机基团或衍生自无机材料的基团可具有反应性基团。在这种情况下,另一种物质可以通过反应性基团进一步连接。反应性基团的实例包括乙炔基、亚乙炔基、乙烯基、叠氮基、环氧基、醛、氧基氨基、卤素等。已知乙炔基和亚乙炔基各自与叠氮基发生1,3
‑
偶极环
加成反应形成1,2,3
‑
三唑环。乙烯基与硫醇基反应形成键。环氧基与氨基或硫醇基反应形成键。醛基与氨基反应形成席夫碱,席夫碱被还原形成键。氧基氨基与酮基或醛基反应形成肟。已知叠氮基与炔基发生1,3
‑
偶极环加成反应形成1,2,3
‑
三唑环。
[0141]
r3表示氢原子、有机基团或衍生自无机材料的基团。有机基团和无机材料的实例包括上述作为由r4表示的有机基团和无机材料衍生基团的实例的那些。当r3为有机基团或无机材料衍生的基团时,有机基团或无机材料衍生的基团优选具有反应性基团。在这种情况下,另一种物质可以通过反应性基团进一步连接,如上所述。
[0142]
在本发明的一个实施方案中,r3是氢原子。在这种情况下,式(1)例如为式(1a)、(1aa)或(1ba):
[0143][0144]
其中r1、r2和r4定义如上。
[0145]
由式(1)表示的化合物包括立体异构体和旋光异构体,并且这些异构体没有特别限制。
[0146]
由式(1)表示的化合物的盐没有特别限制。盐可以是酸式盐或碱式盐。酸式盐的实例包括无机酸盐,例如盐酸盐、氢溴酸盐、硫酸盐、硝酸盐、高氯酸盐和磷酸盐;和有机酸盐,例如醋酸盐、丙酸盐、酒石酸盐、延胡索酸盐、马来酸盐、苹果酸盐、柠檬酸盐、甲磺酸盐和对甲苯磺酸盐。碱式盐的实例包括碱金属盐,例如钠盐和钾盐;碱土金属盐,例如钙盐和镁盐;氨盐;有机胺盐,例如吗啉、哌啶、吡咯烷、单烷基胺、二烷基胺、三烷基胺、单(羟烷基)胺、二(羟烷基)胺和三(羟烷基)胺等。
[0147]
式(1)表示的化合物也可以是水合物或溶剂合物。溶剂的实例包括有机溶剂(例如乙醇、甘油和乙酸)等。
[0148]1‑
2.合成方法1
[0149]
式(1)表示的化合物可以通过各种方法合成。例如,式(1)表示的化合物可以通过包括使式(2)表示的化合物或其盐与式(3)表示的化合物、式(4)表示的化合物或式(9)表示的化合物反应的方法合成,
[0150]
r4‑
r5(2),
[0151]
其中r4如上所定义;r5表示
‑
n3、
‑
x(其中x表示卤素原子)、
‑
b(oh)2、
‑
b(or
51
)2(其中每个r
51
相同或不同并表示一个烃基,条件是两个r
51
与相邻的氧原子可以形成一个环),或
‑
n
2+
,
[0152][0153]
其中r3如上所定义;r6、r7和r8相同或不同,均表示烷基;y表示反应性基团。
[0154]
x表示的卤素原子的例子包括氟、氯、溴、碘等。优选地,由x表示的卤素原子是例如溴。
[0155]
由r
51
表示的烃基的实例包括烷基、环烷基等。
[0156]
由r
51
表示的烷基包括直链烷基和支链烷基。烷基中的碳原子数没有特别限制,例如为1至8个。烷基的具体实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、新戊基、正己基、3
‑
甲基戊基、正庚基、正辛基等。
[0157]
r
51
表示的环烷基中的碳原子数没有特别限制,例如为3至10,优选为4至10。环烷基的具体实例包括环丙基、环丁基、环戊基、环己基、环庚基、环辛基等。
[0158]
由两个r
51
与相邻的氧原子一起形成的环可以被烷基等取代。
[0159]
r6、r7、r8所表示的烷基通常采用直链低级烷基,优选采用乙基。
[0160]
由y表示的反应性基团的实例包括上述作为可包含在有机基团或衍生自无机材料的基团中的反应性基团的实例的那些。y优选为卤素原子(y’)。
[0161]
下面描述了每种所用材料的详细信息。
[0162]1‑2‑
1.情况1(r5为
‑
n3,使用式(3)表示的化合物的情况)
[0163]
就产率等而言,相对于每摩尔式(3)表示的化合物,式(2)表示的化合物的用量,以待反应的官能团(在这种情况下为叠氮基)的摩尔数计,通常优选为0.1至5摩尔,更优选为0.3至2摩尔。
[0164]
该反应通常在反应溶剂的存在下进行。反应溶剂的例子包括,但不特别限于水、甲醇、四氢呋喃、二恶烷、二甲亚砜等。这些溶剂可以单独使用,或以两种或更多种的组合使用。优选向溶剂中加入缓冲液,例如磷酸盐缓冲液。当使用水时,该反应的ph值优选接近中性,特别优选6至8.5,更优选6.5至8,甚至更优选7至7.5。
[0165]
该反应优选在合适的催化剂存在下进行。催化剂是例如铜催化剂。铜催化剂的实例包括二价铜,例如硫酸铜;一价铜,如碘化铜;之类的。此外,还原剂(例如对苯二酚或抗坏血酸钠)、配体等也可用于该反应。
[0166]
就产率等而言,相对于每摩尔本发明的含叠氮基的蛋白质或肽,铜催化剂的用量通常优选为0.1至5摩尔。
[0167]
除了上述组分之外,在该反应中可以适当地使用添加剂,只要不显著损害反应的进程即可。
[0168]
反应可在加热、室温或冷却下进行;并且通常优选在本发明的含叠氮基的蛋白质或肽不显著变性的温度下进行,例如0至45℃(特别是0至40℃)。反应时间没有特别限制,通
常可以为30分钟至3小时,特别是1至2小时。
[0169]
反应进程可以通过色谱法或其他常用方法监测。反应完成后,蒸除溶剂,产物可根据需要通过层析、重结晶或其他常用方法进行分离和纯化。产物的结构可以通过元素分析、ms(esi
‑
ms)分析、ir分析、1h
‑
nmr、
13
c
‑
nmr等进行鉴定。
[0170]1‑2‑
2.情况2(r5为
‑
n3以外的基团,使用式(3)表示的化合物的情况)
[0171]
这种情况与情况1相同,只是在反应体系中加入了用于将r5转化为叠氮基的叠氮化剂。
[0172]
叠氮化剂的实例包括无机叠氮化物,例如叠氮化钠;磺酰叠氮化物;甲硅烷基叠氮化物;磷酰叠氮化物;烷基铵叠氮化物;之类的。
[0173]
就产率等而言,相对于每摩尔式(2)表示的化合物,叠氮化剂的用量通常优选为0.1至5摩尔,更优选为0.3至2摩尔。
[0174]1‑2‑
3.情况3(使用式(4)表示的化合物的情况)
[0175]
在这种情况下,式(1)表示的化合物可以在式(2)表示的化合物与式(4)表示的化合物反应后通过几个步骤合成。例如,在这种情况下,由式(1)表示的化合物可以根据以下方案合成。
[0176][0177]
其中r1、r2、r3、r4、r5和r8如上所定义。
[0178]
步骤1
[0179]
就产率等而言,相对于每摩尔式(4)表示的化合物,式(2)表示的化合物的用量,以待反应的官能团(此时为叠氮基)的摩尔数计,通常优选为0.1~5摩尔,更优选为0.3~2摩尔。
[0180]
使用的溶剂没有限制,只要它不会对反应产生不利影响。实例包括水、醇基溶剂(例如甲醇、乙醇、异丙醇、正丁醇、三氟乙醇和乙二醇)、酮基溶剂(例如丙酮和甲乙酮)、醚基溶剂(例如四氢呋喃、二恶烷、乙醚、二甲氧基乙烷和二甘醇二甲醚)、酯基溶剂(例如乙酸甲酯和乙酸乙酯)、非质子极性溶剂(例如乙腈、n,n
‑
二甲基甲酰胺和二甲亚砜)、卤代烃类溶剂(例如,二氯甲烷和二氯乙烷)及其混合物。溶剂优选为水和非质子极性溶剂(特别是二甲基甲酰胺)的混合物。
[0181]
步骤1通常在碱存在下进行。碱可以是例如有机碱。有机碱的的例子包括三烷基胺(例如三甲胺、三乙胺和n,n
‑
二异丙基乙胺)、吡啶、喹啉、哌啶、咪唑、甲基吡啶、4
‑
二甲氨基吡啶、n,n
‑
二甲基苯胺、n
‑
甲基吗啉、1,5
‑
二氮杂双环[4.3.0]壬
‑5‑
烯(dbn)、1,4
‑
二氮杂双
环[2.2.2]辛烷(dabco)、1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯(dbu)等。当这些碱为液体形式时,它们也可用作溶剂。这些碱可以单独使用,也可以两种以上组合使用。碱优选为哌啶。
[0182]
当使用碱时,相对于每摩尔式(4)表示的化合物,碱的量通常为0.05至1摩尔,优选0.1至0.5摩尔。
[0183]
反应温度没有特别限制。反应通常在冷却、室温或加热下进行。反应可以在优选约50至100℃,更优选约70℃至90℃的温度下进行1至30小时。
[0184]
反应完成后,蒸除溶剂,产物可用层析、重结晶或其他常用方法分离纯化。产物的结构可以通过元素分析、ms(esi
‑
ms)分析、ir分析、1h
‑
nmr、
13
c
‑
nmr等进行鉴定。
[0185]
步骤2
[0186]
步骤2中,式(1”)表示的化合物在还原剂和适量的碱的存在下反应。
[0187]
还原剂的实例包括硼氢化钠、硼氢化锌(zn(bh4)2)、三乙酰氧基硼氢化四甲基铵、三仲丁基硼氢化锂、硼烷、四氢呋喃硼烷络合物、硼烷
‑
二甲基硫醚络合物、氢化铝锂、二异丁基氢化铝、硼氢化锂,之类的。这些还原剂可以单独使用,也可以两种以上组合使用。还原剂优选为硼氢化钠。
[0188]
相对于每摩尔式(1”)表示的化合物,还原剂的用量通常为1至15摩尔,优选3至10摩尔。
[0189]
作为碱,例如,可以使用无机碱。无机碱的实例包括碱金属(例如钠和钾)、碱金属碳酸氢盐(例如碳酸氢锂、碳酸氢钠和碳酸氢钾)、碱金属氢氧化物(例如氢氧化锂、氢氧化钠、氢氧化钾和氢氧化铯)、碱金属碳酸盐(例如碳酸锂、碳酸钠、碳酸钾和碳酸铯)、碱金属低级(c1‑
c4)醇盐(例如甲醇钠、乙醇钠和叔丁醇钾)、碱金属氢化物(例如氢化钠和氢化钾)等。这些碱可以单独使用,也可以两种以上组合使用。碱优选为碱金属低级(c1‑
c4)醇盐(特别是甲醇钠)。
[0190]
使用的溶剂没有限制,只要它不会对反应产生不利影响。实例包括水、醇基溶剂(例如甲醇、乙醇、异丙醇、正丁醇、三氟乙醇和乙二醇)、酮基溶剂(例如丙酮和甲乙酮)、醚基溶剂(例如四氢呋喃)、二恶烷、乙醚、二甲氧基乙烷和二甘醇二甲醚)、酯基溶剂(例如乙酸甲酯和乙酸乙酯)、非质子极性溶剂(例如乙腈、n,n
‑
二甲基甲酰胺和二甲亚砜)、卤代烃类溶剂(例如,二氯甲烷和二氯乙烷),以及它们的混合物。溶剂优选醇基溶剂(特别是甲醇)。
[0191]
反应温度没有特别限制。反应通常在冷却、室温或加热下进行。可以在优选约0℃至60℃、更优选约10℃至40℃的温度下进行1至30小时。
[0192]
反应完成后,蒸除溶剂,产物可用层析、重结晶或其他常用方法分离纯化。产物的结构可以通过元素分析、ms(esi
‑
ms)分析、ir分析、1h
‑
nmr、
13
c
‑
nmr等进行鉴定。
[0193]
步骤3
[0194]
在步骤3中,式(1’)表示的化合物在氧化剂的存在下反应。
[0195]
作为氧化剂,例如可以使用二氧化锰。可用氧化剂的其他实例包括二氧化硒、硝酰基自由基例如2,2,6,6
‑
四甲基哌啶1
‑
氧基(tempo)和2
‑
氮杂金刚烷
‑
n
‑
氧基(azado)等。这些氧化剂可以单独使用,也可以两种以上组合使用。
[0196]
相对于每摩尔式(1’)表示的化合物,氧化剂的用量通常为3至20摩尔,优选5至15摩尔。
[0197]
使用的溶剂没有限制,只要它不会对反应产生不利影响。实例包括酮基溶剂(例如,丙酮和甲乙酮)、醚基溶剂(例如,四氢呋喃、二恶烷、乙醚、二甲氧基乙烷和二甘醇二甲醚)、酯基溶剂(例如,乙酸甲酯和乙酸乙酯)、非质子极性溶剂(例如、乙腈、n,n
‑
二甲基甲酰胺和二甲亚砜)、卤代烃类溶剂(例如氯仿、二氯甲烷和氯乙烯),以及它们的混合物。溶剂优选为卤代烃类溶剂(特别是氯仿)。
[0198]
反应温度没有特别限制。反应通常在冷却、室温或加热下进行。反应可以在优选约0℃至60℃,更优选约10℃至40℃的温度下进行1至30小时。
[0199]
反应完成后,蒸除溶剂,产物可用层析、重结晶或其他常用方法分离纯化。产物的结构可以通过元素分析、ms(esi
‑
ms)分析、ir分析、1h
‑
nmr、
13
c
‑
nmr等进行鉴定。
[0200]1‑2‑
4.情况4(r5为
‑
n3,使用式(9)表示的化合物的情况)
[0201]
在这种情况下,式(1)表示的化合物可以在式(2)表示的化合物与式(9)表示的化合物反应后通过几个步骤合成。例如,在这种情况下,由式(1)表示的化合物可以根据以下方案合成。
[0202][0203]
可以根据或基于上述情况1来执行步骤4。可以根据或基于上述步骤3来执行步骤6。步骤5可以通过使式(1**)表示的化合物与式(10)表示的化合物:r3‑
z反应来进行。z是与y反应的反应性基团。当y为卤素时,例如可以采用
‑
b(oh)2作为z。步骤5的反应条件可以根据已知信息,根据y和z的类型确定。
[0204]1‑
3.合成方法2
[0205]
除了上述合成方法1之外,式(1)表示的化合物可以通过例如包括使式(5)表示的化合物:r4‑
r9(5),其中r4如上所定义;r9表示
‑
nr
9a
r
9b
(其中r
9a
和r
9b
相同或不同,各自表示一个氢原子或烷基)与式(6)表示的化合物反应的方法来合成:
[0206][0207]
其中r
10
表示吸电子基团;r
11
表示
‑
r
11a
‑
r
12
(其中r
11a
表示单键或连接体;且r
12
表示载体);n表示0或1;且m表示1~5的整数。
[0208]
此外,式(1aa)表示的化合物可以通过使用式(5’)表示的化合物合成:r9‑
r
4a
‑
r9(5’),其中r
4a
和r
5a
如上所定义,作为由式(5)表示的化合物。
[0209]
烷基的实例包括但不特别限于低级烷基,例如甲基和乙基。r
9a
和r
9b
均优选为氢原子。
[0210]
吸电子基团的实例包括但不特别限于
‑
no2、
‑
f、cf3、
‑
cn、
‑
coome等。吸电子基团的位置优选为对位,例如在
‑
no2的情况下,并且优选邻位和间位,例如在
‑
f和cf3的情况下。在后一种情况下,更优选地,
‑
cn位于对位。
[0211]
当吸电子基团为例如
‑
no2时,m优选为1。当吸电子基团为例如
‑
f或cf3时,m优选为4至5,更优选所有邻位和间位被替换为r
10
。在后一种情况下,甚至更优选地,
‑
cn位于对位。
[0212]
当
‑
f或cf3(优选
‑
f)在所有邻位和间位,及
‑
cn在对位时,与式(5)表示的化合物的反应即使在相对低温度条件下(优选20
‑
60℃,更优选为25
‑
40℃)也能充分进行。
[0213]
连接体没有特别限制,只要它能够将载体连接到苯环上即可。实例包括在主链中包含任何以下部分结构的连接体。
[0214][0215]
构成连接体主链的原子数为例如1至100、1至50、1至20或1至10。
[0216]
载体没有特别限制。
[0217]
载体粒子的平均粒径没有特别限定,优选为在溶液中容易沉淀的大小。载体粒子的平均粒径例如为1nm~1mm,优选为10nm~100μm。
[0218]
载体粒子的材质没有特别限定,例如金、银、铜、铁、铝、镍、锰、钛等金属粒子及其氧化物;树脂粒子,如聚苯乙烯和乳胶;二氧化硅粒子;之类的。载体粒子的形状没有特别限定,例如可以是球体、长方体、立方体、三棱锥等形状。载体粒子可以在表面上具有使另一种物质(例如,结合物质2)的结合更容易和/或更强的物质。所述物质的实例包括含反应基团的物质,例如含环氧基团的物质、含氨基的物质、含羧基的物质、含叠氮基的物质;对其他分子具有亲和力的物质,例如亲和素、蛋白质a和蛋白质b;之类的。载体粒子可以进一步包含标记物质。可以仅使用一种类型的载体粒子,或者可以组合使用两种或更多种类型的载体粒子。
[0219]
就产率等而言,相对于每摩尔由式(6)表示的化合物,由式(5)表示的化合物的使用量通常优选为0.1至5摩尔,并且更优选为0.3至2摩尔。
[0220]
使用的溶剂没有限制,只要它不会对反应产生不利影响。实例包括水、醇基溶剂(例如甲醇、乙醇、异丙醇、正丁醇、三氟乙醇和乙二醇)、酮基溶剂(例如丙酮和甲乙酮)、醚基溶剂(例如四氢呋喃、二恶烷、乙醚、二甲氧基乙烷和二甘醇二甲醚)、酯基溶剂(例如乙酸甲酯和乙酸乙酯)、非质子极性溶剂(例如乙腈、n,n
‑
二甲基甲酰胺和二甲亚砜)、卤代烃类溶剂(例如二氯甲烷和二氯乙烷)及其混合物。溶剂优选为水和醇类溶剂的混合物。
[0221]
在该反应中,从产率等方面考虑,优选使用酸催化剂。酸催化剂的实例包括但不特别限于乙酸、甲磺酸、对甲苯磺酸、2
‑
吗啉乙磺酸(mes)、3
‑
吗啉丙磺酸(mops)、4
‑
(2
‑
羟乙基)
‑1‑
哌嗪乙磺酸(hepes)、n
‑
三(羟甲基)甲基
‑3‑
氨基丙磺酸(taps)等。其中,从进一步不妨碍后续的蛋白质修饰反应的观点考虑,优选mes、mops、hepes、taps等古德缓冲液。
[0222]
反应温度没有特别限制。反应通常在冷却、室温或加热下进行。反应可优选在约20℃至100℃的温度下进行10分钟至30小时。
[0223]
反应完成后,蒸除溶剂,产物可用层析、重结晶或其他常用方法分离纯化。产物的结构可以通过元素分析、ms(esi
‑
ms)分析、ir分析、1h
‑
nmr、
13
c
‑
nmr等进行鉴定。
[0224]
当式(6)表示的化合物含有载体时(即,当r
11
为
‑
r
11a
‑
r
12
时),纯化操作可以以简单的方式进行。具体地,式(6x)表示的化合物,其为反应产生的副产物(苯胺衍生物):
[0225][0226]
其中,r
9a
、r
9b
、r
10
、r
11a
、r
12
和m的定义如上,可以通过使用载体等的沉淀操作容易地去除。因此,在本发明的一个实施方案中,本发明涉及由式(6’)表示的化合物:
[0227][0228]
其中r
10
、r
11
、r
12
和m如上所定义。
[0229]1‑
4.用途
[0230]
本发明的修饰分子可用于将另一个分子/物质连接到蛋白质或肽的n端,例如,生产后述的本发明的复合物质(式(7)表示的化合物或其盐,或该化合物或其盐的水合物或溶剂合物)。因此,本发明的修饰分子可以适合用作试剂,特别是用作蛋白质和/或肽修饰试剂的活性成分。该试剂只要含有本发明的修饰分子就没有特别限定,根据需要还可以含有其他成分。其他成分只要是药学上可接受的成分就没有特别限制。其他成分的实例包括碱、载体、溶剂、分散剂、乳化剂、缓冲剂、稳定剂、赋形剂、粘合剂、崩解剂、润滑剂、增稠剂、湿润剂、着色剂、香料、螯合剂等。
[0231]
2.复合物质
[0232]
在本发明的一个实施方案中,本发明涉及式(7)所表示的化合物或其盐,或者该化合物或其盐的水合物或溶剂合物(在本说明书中,这些可以统称为“本发明的复合物质”):
[0233][0234]
其中r1、r2、r3和r4如上所定义;r
13
表示其中n端氨基酸残基和与其相邻的
‑
nh
‑
被排除在蛋白质或肽之外的基团;和r
14
表示蛋白质或肽的n端氨基酸残基的侧链。下面描述本发明的复合物质。
[0235]
r
13
表示其中n端氨基酸残基和与其相邻的
‑
nh
‑
被排除在蛋白质或肽之外的基团。
[0236]
蛋白质或肽没有特别限制,只要其是n端氨基未修饰并且n端的第二个氨基酸残基是脯氨酸以外的氨基酸残基的蛋白质或肽。在蛋白质或肽中,可以在除n端以外的位点(例如,半胱氨酸残基)进行各种修饰。这种蛋白质或肽的实例包括由式(7a)表示的蛋白质或肽。
[0237][0238]
r
14
表示蛋白质或肽的n端氨基酸残基的侧链。氨基酸残基可以是天然氨基酸残基或合成氨基酸残基。实例包括具有碱性侧链的氨基酸残基,例如赖氨酸、精氨酸和组氨酸;具有酸性侧链的氨基酸残基,如天冬氨酸和谷氨酸;具有不带电荷的极性侧链的氨基酸残基,如甘氨酸、天冬酰胺、谷氨酰胺、丝氨酸、苏氨酸、酪氨酸和半胱氨酸;具有非极性侧链的氨基酸残基,如丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、蛋氨酸和色氨酸;具有β
‑
支链侧链的氨基酸残基,如苏氨酸、缬氨酸、异亮氨酸;具有芳香侧链的氨基酸残基,如酪氨酸、苯丙氨酸、色氨酸和组氨酸;之类的。
[0239]
蛋白质或肽没有特别限制并且可以是天然的、合成的或人工的。
[0240]
蛋白质或肽可以被化学修饰。蛋白质或肽的c端可以是羧基(
‑
cooh)、羧酸盐(
‑
coo
‑
)、酰胺(
‑
conh2)或酯(
‑
coor)中的任一个。此处,酯中的r例如为甲基、乙基、正丙基、异
丙基或正丁基等c1‑6烷基;例如,c3‑8环烷基,例如环戊基和环己基;例如,c6‑
12
芳基,例如苯基和α
‑
萘基;例如,苯基
‑
c1‑2烷基,如苄基和苯乙基;包括α
‑
萘基
‑
c1‑2烷基的c7
‑
14芳烷基,例如α
‑
萘基甲基;新戊酰氧基甲基等。在蛋白质或肽中,除c端之外的羧基(或羧酸盐)可以被酰胺化或酯化。作为这种情况下的酯,例如,使用上述c端酯等。此外,蛋白质或肽包括其中分子中氨基酸侧链中的取代基(例如,
‑
oh、
‑
sh、氨基、咪唑基、吲哚基或胍基)被合适的保护基团(例如,包括c1‑6烷酰基的c1‑6酰基,例如甲酰基或乙酰基)保护的蛋白质或肽。
[0241]
蛋白质或肽可以被翻译后修饰,或者可以通过人工酶处理或化学修饰被翻译后修饰。翻译后修饰的实例包括磷酸化、n
‑
糖基化、o
‑
糖基化、c
‑
糖基化、磷酸糖基化、糖基磷脂酰肌醇化、s
‑
亚硝基化、甲基化、n
‑
乙酰化、s
‑
肉豆蔻酰化、s
‑
异戊二烯化、s
‑
棕榈酰化等。蛋白质或肽可以是添加了蛋白质或肽,例如已知的蛋白质标签或信号序列,或标记物质的蛋白质或肽。蛋白质标签的例子包括生物素、his标签、flag标签、halo标签、mbp标签、ha标签、myc标签、v5标签、pa标签、spy标签等。信号序列的例子包括核定位信号等。
[0242]
蛋白质或肽可以单独作为单个分子存在,或者可以与另一个分子连接形成复合物。例如,蛋白质或肽可以是存在于细胞表面的蛋白质或肽、细胞破碎液中的蛋白质或肽、或负载在某些物质上的蛋白质或肽。连接的方式没有特别限定,例如可以是氢键、静电力、范德华力、疏水键、共价键、配位键等。
[0243]
只要存在n端,蛋白质或肽就可以被酶或化学反应断裂。当蛋白质或肽被n端修饰时,n端可以通过酶或化学反应去修饰。
[0244]
式(7)表示的化合物包括立体异构体和旋光异构体,对这些异构体没有特别限制。
[0245]
由式(7)表示的化合物的盐没有特别限制。盐可以是酸式盐或碱式盐。酸式盐的实例包括无机酸盐,例如盐酸盐、氢溴酸盐、硫酸盐、硝酸盐、高氯酸盐和磷酸盐;和有机酸盐,例如乙酸盐、丙酸盐、酒石酸盐、延胡索酸盐、马来酸盐、苹果酸盐、柠檬酸盐、甲磺酸盐和对甲苯磺酸盐。碱式盐的实例包括碱金属盐,例如钠盐和钾盐;碱土金属盐,如钙盐和镁盐;氨盐;有机胺盐,例如吗啉、哌啶、吡咯烷、单烷基胺、二烷基胺、三烷基胺、单(羟烷基)胺、二(羟烷基)胺和三(羟烷基)胺等。
[0246]
式(7)表示的化合物也可以是水合物或溶剂合物。溶剂的实例包括有机溶剂(例如乙醇、甘油和乙酸)等。
[0247]
式(7)表示的化合物可以通过各种方法合成。例如,式(7)表示的化合物可以通过包括使蛋白质或肽与本发明的修饰分子反应的方法来生产。该反应可以在本发明的修饰分子的合成反应(特别优选上面“1
‑
2.合成方法2”反应)之后进行。在这种情况下,例如,可以将本发明的修饰分子的合成反应中得到的反应混合物用该反应中使用的溶剂稀释,然后进行该反应。
[0248]
就产率等而言,相对于每摩尔蛋白质或肽,本发明的修饰分子的使用量通常优选为5~400摩尔。
[0249]
该反应通常在反应溶剂的存在下进行。反应溶剂的实例包括但不特别限于水等。这些溶剂可以单独使用,或以两种或更多种的组合使用。此外,优选在溶剂中加入缓冲液,例如磷酸盐缓冲液。当使用水时,就n端选择性而言,该反应的ph优选接近中性,特别优选6至8.5,更优选6.5至8,甚至更优选7至7.5。
[0250]
除了上述组分之外,在该反应中可以适当地使用添加剂,只要不显著损害反应进
程即可。
[0251]
反应可以在加热、室温或冷却下进行。反应通常优选在蛋白质或肽不会显著变性的温度下进行,例如0℃至45℃(特别是0℃至40℃)。反应时间没有特别限制,通常可以为8至36小时,特别是12至24小时。
[0252]
反应进程可以通过层析或其他常用方法监测。反应完成后,蒸除溶剂,产物可根据需要通过层析、重结晶或其他常用方法进行分离和纯化。产物的结构可以通过元素分析、ms(esi
‑
ms)分析、ir分析、1h
‑
nmr、
13
c
‑
nmr等进行鉴定。
[0253]
当式(1aa)表示的化合物用作本发明的修饰分子时,式(7aa)表示的化合物由上述反应获得:
[0254][0255]
其中r3、r
4a
、r
13
和r
14
如上定义。在这种情况下,有机分子等可以通过与羟胺进一步形成肟而连接。具体地,式(1aa)表示的化合物可以与式(8)表示的化合物反应:r4‑
o
‑
nh2(8),其中r4如上定义,以合成式(7aaa)表示的化合物:
[0256][0257]
其中r3、r
4a
、r4、r
13
和r
14
如上所定义。该合成反应的条件等可以根据或基于从醛开始与羟胺形成肟的已知反应条件来确定。该反应也适用于蛋白质修饰,因为它甚至在温和条件下的水中也能进行。
[0258]
本发明的复合物质具有在蛋白质或肽上连接有其他物质的结构。在取决于要连接的物质的各个领域中,本发明的复合物质可用作例如抗体
‑
药物缀合物、标记蛋白质试剂、蛋白质固定无机材料、其中蛋白质是连接的融合蛋白,或具有与其融合的核酸的蛋白质。
[0259]
在本发明的复合物质的一个实施方案中,复合物质可以设计成与蛋白质或肽连接的物质逐渐解离。因此,在将例如药物与蛋白质或肽连接的本发明的复合物质施用于生物体后,通过利用在生物体内本发明的复合物质的药物的逐渐解离,药物可以在生物体内逐渐释放。
[0260]
当本发明的复合物质具有反应性基团时(例如,当r3具有反应性基团时),反应性基团可用于进一步连接另一物质。例如,它可用于通过使用叠氮基团的反应(例如,huisgen环加成反应、应变促进的叠氮化物
‑
炔环加成反应,或staudinger
‑
bertozzi连接)连接其他物质(例如有机分子、有机分子复合物或无机材料)。
[0261]
实施例
[0262]
下面给出实施例以更详细地说明本发明;然而,本发明不限于这些实施例。
[0263]
实施例1.化合物合成1(方法a)
[0264]1‑
1.使用的设备
[0265]
使用bruker avance iii hd核磁共振光谱仪测量核磁共振(nmr)谱,并使用测量溶剂的剩余信号作为内参计算化学位移。使用bruker microtof focus iii质谱仪进行电喷雾电离飞行时间质谱(esi
‑
tof ms),并使用甲醇或乙腈(均为hplc级)作为流动相。傅里叶变换红外吸收(ft
‑
ir)光谱使用jasco ft/ir
‑
4000傅里叶变换红外分光光度计在atr模式下使用金刚石或镓棱镜测量。
[0266]1‑
2.试剂、溶剂等
[0267]
合成中使用的试剂和溶剂直接使用市售品。所使用的前体叠氮化物是参考已发表的报告合成的(y.zhao,p.gong,bioorg.med.chem.,2014,22,6438
‑
6452)。
[0268]1‑3‑
1.1
‑
(3
‑
羧基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(1)的合成
[0269]
根据以下方案合成化合物1。
[0270][0271]
参考已发表的报告(ta bakka,mb strom,jh anderson,or gautun,bioorg.med.chem.lett.,2017,27,1119
‑
1123,and jt fletcher,ja cristensen,em villra,tetrahedron lett.,2017,58,4450
‑
4454)合成化合物1。具体合成过程和化合物鉴定结果如下所述。
[0272]
在氮气氛下,将叠氮化物(0.60mmol)、炔丙醛二乙缩醛(115μl,0.80mmol)和抗坏血酸钠(60mg,0.30mmol)加入五水合硫酸铜(ii)(38mg,0.15mmol)的水溶液(7.5ml)和叔丁醇(7.5ml)的混合物中,将混合物在70℃下搅拌24小时,并在室温下在空气中搅拌1小时。所得悬浮液用饱和氯化钠水溶液(10ml)稀释,用乙酸乙酯(50ml
×
3)萃取,并用饱和氯化钠水溶液(20ml
×
2)洗涤。将得到的有机层用硫酸钠干燥,减压蒸馏除去过滤得到的滤液的溶剂。残余物通过硅胶柱层析纯化,得到化合物1(黄色固体)。图1显示了1h nmr谱。
[0273]
产率48%;1h nmr(400mhz,dmso
‑
d6):δ10.12(s,1h),9.67(s,1h),8.48(s,1h),8.16(d,j=7.8hz,1h),8.09(d,j=7.8hz,1h),7.72(t,,j=7.8hz,1h);
13
c nmr(100mhz,dmso
‑
d6):δ184.9,167.0,147.6,136.0,130.1,130.0,126.6,124.0,121.3;esi
‑
tof ms(正模式)计算c
10
h7nan3o3[m+na]+240.04的m/z,求得240.04;ft
‑
ir(atr模式,镓棱镜),νcm
‑1:3128,2923,1697,1263,1230,1184,757,673,647。
[0274]1‑3‑
2.1
‑
(4
‑
羧基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(2)的合成
[0275][0276]
使用4
‑
叠氮基苯甲酸作为前体,使用方法a如实施例1
‑3‑
1合成化合物2(黄色固体)。图2显示了1h nmr谱。
[0277]
产率63%;1h nmr(400mhz,dmso
‑
d6):δ10.13(s,1h),9.69(s,1h),25 8.18
‑
8.12(m,4h);esi
‑
tof ms(正模式)计算c
10
h7nan3o3[m+na]+240.04的m/z,求得240.04;ft
‑
ir(atr模式,镓棱镜),νcm
‑1:3110,1695,1683,1605,1429,1319,1294,1264,989,945,864,773,702,688。
[0278]1‑3‑
3.1
‑
(3,5
‑
二羧基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(3)的合成
[0279][0280]
化合物3(黄色固体)使用5
‑
叠氮间苯二甲酸为前体,通过使用方法a如实施例1
‑3‑
1合成。图3显示了1h nmr谱。
[0281]
产率10%;1h nmr(400mhz,dmso
‑
d6):δ10.12(s,1h),9.81(s,1h),8.65(s,2h),8.56(s,1h);ft
‑
ir(atr模式,镓棱镜),νcm
‑1:3133,1697,1296,1281,1247,1049,830,757,678,666,566,527。
[0282]1‑3‑
4.1
‑
(4
‑
(二乙氨基)苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(4)的合成
[0283][0284]
使用4
‑
叠氮基
‑
n,n
‑
二乙基苯胺作为前体,使用方法a如实施例1
‑3‑
1合成化合物4(棕色固体)。图4显示了1h nmr谱。
[0285]
产率60%;1h nmr(400mhz,cdcl3):δ10.12(s,1h),8.36(s,1h),7.52(d,j=
9.1hz,2h),6.73(d,j=9.1hz,2h),3.42(q,j=7.1hz,4h),1.21(t,j=7.1hz,6h);
13
c nmr(100mhz,dmso
‑
d6):δ185.0,148.0,147.3,125.1,124.3,122.1,111.4,43.8,12.3;esi
‑
tof ms(正模式)计算c
13
h
16
nan4o[m+na]+267.12的m/z,求得267.13。
[0286]1‑3‑
5.1
‑
(4
‑
甲氧基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(5)的合成
[0287][0288]
使用1
‑
叠氮基
‑4‑
甲氧基苯作为前体通过使用方法a如实施例1
‑3‑
1中合成化合物5(白色固体)。图5显示了1h nmr谱。
[0289]
产率68%;1h nmr(400mhz,dmso
‑
d6):δ10.12(s,1h),8.43(s,1h),7.66(d,j=8.9hz,2h),7.06(d,j=8.9hz,2h),3.89(s,3h);
13
c nmr(100mhz,dmso
‑
d6):δ185.0,159.9,147.5,129.3,125.9,122.4,115.0,55.7;esi
‑
tof ms(正模式)计算c
10
h9nan3o
‑2[m+na]+226.06的m/z,求得226.06。
[0290]1‑3‑
6.1
‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(6)的合成
[0291][0292]
使用叠氮苯作为前体通过使用方法a如实施例1
‑3‑
1中合成化合物6(棕色固体)。图6显示了1h nmr谱。
[0293]
产率9%;1h nmr(400mhz,dmso
‑
d
6)
:δ10.11(s,1h),9.57(s,1h),7.97(d,j=8.0hz,2h),7.66
‑
7.54(m,3h).
[0294]
实施例2.化合物合成2(方法b)
[0295]
所用设备、试剂、溶剂等与实施例1类似。
[0296]2‑
1.1
‑
苄基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(7)的合成
[0297][0298]
化合物7是参考已发表的报告合成的(j.t.fletcher,tetrahedron lett.,2017,58,4450
‑
4454)。具体合成过程和化合物鉴定结果如下所述。
[0299]
将五水合硫酸铜20(ii)(47.8mg,0.19mmol)的水溶液(7.5ml)和叔丁醇(7.5ml)的混合物冷却至0℃,在氮气氛下向混合物中加入叠氮化钠(103mg,1.6mmol))。室温搅拌10分钟后,向混合物中加入溴化苄(179μl,1.5mmol)和炔丙醛二乙缩醛(240μl,1.7mmol),并将
nmr谱。
[0311]
产率71%;1h nmr(400mhz,cdcl3):δ10.2(s,1h),8.48(s,1h),7.64(d,j=8.3hz,2h),7.37(d,j=8.3hz,2h),2.45(s,3h);
13
c nmr(100mhz,cdcl3):δ185.3,148.2,140.3,134.0,130.7,123.2,120.9,21.3;esi
‑
tof ms(正模式)计算c
10
h9n3ona[m+na]
+
210.06m/z,求得210.06。
[0312]
实施例4.化合物合成4(方法d)
[0313]
所用设备、试剂、溶剂等与实施例1类似。
[0314]4‑
1.1
‑
(4
‑
硝基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(10)的合成
[0315][0316]
化合物10的合成参考了已发表的报告(j.t.fletcher,j.e.reilly,tetrahedron lett.,2011,52,5512
‑
5515)。具体合成过程和化合物鉴定结果如下所述。
[0317]
将叠氮化钠(358mg,5.5mmol)的水溶液(25ml)和叔丁醇(25ml)的混合物冷却至0℃,并分批加入硝基苯重氮四氟硼酸盐(1.18g,5.0mmol),然后剧烈搅拌1小时。随后,在氮气氛下加入炔丙醛二乙缩醛(783μl,5.5mmol)和抗坏血酸钠(396mg,2mmol),然后在70℃下搅拌过夜。过滤反应溶液,然后用乙酸乙酯(50ml
×
3)萃取滤液。有机层经硫酸钠干燥,减压蒸馏除去溶剂。所得粗产物通过再沉淀(己烷:乙酸乙酯)纯化,得到化合物10(黄色固体)。图10显示了1h nmr谱。
[0318]
产率62%;1h nmr(400mhz,dmso
‑
d6):δ10.15(s,1h),9.78(s,1h),8.49(d,j=9.2hz,2h),8.32(d,j=9.2hz,2h);
13
c nmr(100mhz,dmso
‑
d6):δ185.0,147.8,147.4,140.3,126.9,125.6,121.5;esi
‑
tof ms(正模式)计算c9h6n4o3na[m+na]
+
241.03m/z,求得241.03.
[0319]
实施例5.化合物合成5(方法e)
[0320]
所用设备、试剂、溶剂等与实施例1类似。
[0321]5‑
1.5
‑
甲基
‑1‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(13)的合成
[0322]
化合物13的合成参考了已发表的报告(j.zhang,g.jin,s.xiao,j.wu,s.guo,tetrahedron 2013,69,2352
‑
2356;i.ibnusaud,b.singaram,j.org.chem.,2018,83,1431
‑
1440;和t.ismail,s.shafi,i.hyder,t.sidiq,a.khajuria,smalam,msk halmuthur,arch.pharm.chem.生命科学,2015,348,796
‑
807)。
[0323][0324]5‑1‑
1.5
‑
甲基
‑1‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
羧酸乙酯(11)的合成
[0325]
将乙酰乙酸乙酯(1.5mmol)和哌啶(20μl,0.2mmol)添加到叠氮化苯(120mg,1.0mmol)在二甲基甲酰胺(10ml)和纯水(1ml)的混合物中的溶液中,并将混合物在80℃搅拌24小时。向所得溶液中加入水(20ml)以停止反应,随后用乙醚(20ml
×
5)萃取。所得有机层用水(20ml
×
2)洗涤。将有机层用硫酸钠干燥,减压蒸馏除去过滤得到的滤液的溶剂。所得粗产物通过硅胶柱层析(己烷:乙酸乙酯)纯化,得到化合物11(白色固体)。
[0326]
产率27%;1h nmr(400mhz,cdcl3):δ7.61
‑
7.55(m,3h),7.47
‑
7.44(m,2h),4.47(q,j=7.1hz,2h),2.60(s,3h),1.46(t,j=7.1hz,3h)。
[0327]5‑1‑
2.(5
‑
甲基
‑1‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
基)甲醇(12)的合成在氮气氛下,将硼氢化钠(29mg,0.77mmol)加入到化合物11(0.12mmol)和甲醇(1ml)中的甲醇钠(1mg,2μmol)的溶液中,然后在室温下搅拌3小时。向反应溶液中加入过量的甲醇以停止反应。减压浓缩反应液,残渣用饱和氯化钠水溶液(5ml)稀释,随后用乙醚(20ml
×
3)萃取,并用饱和氯化钠水溶液(5ml
×
2)洗涤。所得有机层经硫酸钠干燥。之后,过滤,将滤液的溶剂减压浓缩得到的粗产物通过硅胶柱层析(己烷:乙酸乙酯=2:1)纯化,得到化合物12(白色固体)。图11显示了1h nmr谱。
[0328]
产率37%;1h nmr(400mhz,cdcl3):δ7.58
‑
7.52(m,3h),7.48
‑
7.46(m,2h),4.83(d,j=6.0hz,2h),2.37(s,2h),1.96(t,j=6.0hz,1h)。
[0329]5‑1‑
3.5
‑
甲基
‑1‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(13)的合成
[0330]
将活化的二氧化锰(141mg,1.6mmol)加入到化合物12(0.16mmol)的氯仿(5ml)溶液中,并将混合物在室温下在氮气气氛下搅拌24小时。过滤反应混合物,并减压蒸馏除去滤液的溶剂,得到化合物13(白色固体)。
[0331]
产率89%;1h nmr(400mhz,dmso
‑
d6):δ7.58
‑
7.52(m,3h),7.48
‑
7.46(m,2h),4.83(d,j=6.0hz,2h),2.37(s,2h),1.96(t,j=6.0hz,1h);
13
c nmr(100mhz,dmso
‑
d6):δ186.1,143.3,138.7,134.7,130.3,129.8,125.3,9.3。
[0332]5‑1‑
4.1
‑
苯基
‑5‑
(三氟甲基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
羧酸乙酯(14)的合成
[0333]
[0334]
以4,4,4
‑
三氟乙酰乙酸乙酯为前体,通过与实施例5
‑1‑
1相同的方法合成化合物14(黄色油状物)。
[0335]
产率35%;1h nmr(400mhz,cdcl3):δ7.63
‑
7.57(m,3h),7.47(d,j=7.3hz,2h),4.51(q,j=7.1hz,2h),1.45(t,j=7.1hz,3h)。
[0336]5‑1‑
5.(1
‑
苯基
‑5‑
(三氟甲基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
基)甲醇(15)的合成
[0337][0338]
以化合物14为前体,通过与实施例5
‑1‑
2相同的方法合成化合物15(白色固体)。
[0339]
产率83%;1h nmr(400mhz,cdcl3):δ7.61
‑
7.55(m,3h),7.47(d,j=7.2hz,2h),4.96(d,j=6.0hz,2h)。
[0340]5‑1‑
6.(1
‑
苯基
‑5‑
(三氟甲基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
基)甲醇(16)的合成
[0341][0342]
以化合物15为前体,通过与实施例5
‑1‑
3相同的方法合成化合物16(白色固体)。图12显示了1h nmr谱。
[0343]
产率75%;1h nmr(400mhz,dmso
‑
d6):δ10.21(s,1h),7.74
‑
7.66(m,5h)。
[0344]
实施例6.化合物合成6(方法f)
[0345]
所用设备、试剂、溶剂等与实施例1类似。
[0346]6‑
1.1
‑
(2
‑
(2
‑
(丙
‑2‑
炔
‑1‑
基氧基)乙氧基)乙基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(17)的合成
[0347][0348]
将化合物10(109mg,0.5mmol)和2
‑
(2
‑
(2
‑
丙炔氧基)乙氧基]乙胺(72μl,0.5mmol)分散在纯水(1ml)和叔丁醇(1ml)的混合物中,并在氮气氛下,将混合物在60℃下搅拌过夜,反应液冷却至0℃后,加入0.1m hcl水溶液(50ml)停止反应,然后用乙酸乙酯(20ml
×
3)萃取,有机层用硫酸钠干燥,减压蒸馏除去溶剂,所得粗品经硅胶柱层析(己烷:乙酸乙酯=0~60%)纯化得到化合物17(黄色油状物)。图13为1h nmr谱图。
[0349]
产率70%;1h nmr(400mhz,cd3cn):δ10.04(s,1h),8.42(s,1h),4.59(t,j=5.1hz,2h),4.11(d,j=2.4hz,2h),3.87(t,j=5.1hz,2h),3.60
‑
3.56(m,4h),2.69(t,j=2.4hz,1h);
13
c nmr(100mhz,cd3cn):δ185.7,148.4,128.6,80.8,75.7,70.6,69.7,69.5,58.7,51.3;esi
‑
tof ms(正模式)计算c
10
h
13
n3o3na[m+na]
+
246.08m/z,求得246.08。
[0350]
实施例7.化合物合成7:功能分子的合成
[0351]
所用设备、试剂、溶剂等与实施例1类似。使用的反应前体为购买或适当合成的。
[0352]7‑
1.n
‑
(2
‑
(2
‑
(2
‑
(2
‑
(4
‑
甲酰基
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)乙氧基)乙氧基)乙氧基)乙基)
‑5‑
((4s)
‑2‑
氧代六氢
‑
1h
‑
噻吩并[3,4
‑
d]咪唑
‑4‑
基)戊酰胺(18)的的合成
[0353][0354]
使用n
‑
(2
‑
(2
‑
(2
‑
(2
‑
叠氮乙氧基)乙氧基)乙氧基)乙基)
‑5‑
((4s)
‑2‑
氧代六氢
‑
1h
‑
噻吩并[3,4
‑
d]咪唑
‑4‑
基)戊酰胺作为前体,使用与实施例1
‑3‑
1相同的方法(方法a)合成化合物18(白色固体)。图14显示了1h nmr谱。
[0355]
产率23%;1h nmr(400mhz,cd3cn+dmso
‑
d6(one drop)):δ10.0(s,1h),8.47(s,1h),6.49(s,1h),5.17(s,1h),4.98(s,1h),4.60(t,j=5.0hz,2h),4.42
‑
4.38(m,1h),4.24
‑
4.21(m,1h),3.88(t,j=5.0hz,2h),3.59
‑
3.55(m,2h),3.54(m,6h),3.45(t,j=5.6hz,2h),3.27(t,j=5.6hz,2h),3.17
‑
3.12(m,1h),2.88(dd,j=5.0,12.6hz,1h),2.63(d,j=5.0,12.6hz,1h),2.12(t,j=7.3hz,2h),1.63
‑
1.49(m,4h),1.41
‑
1.34(m,2h);esi
‑
tof ms(正模式)计算c
21
h
34
n6o6na[m+na]
+
521.1,求得521.1。
[0356]7‑
2.1
‑
(3’,6
’‑
二羟基
‑3‑
氧代
‑
3h
‑
螺[异苯并呋喃
‑
1,9
’‑
呫吨]
‑5‑
基)
‑
1h
‑
1,2,3
‑‑
三唑
‑4‑
甲醛(19)的合成
[0357][0358]
以5
‑
叠氮基
‑3’
,6
’‑
二羟基
‑
3h
‑
螺[异苯并呋喃
‑
1,9'
‑
呫吨]
‑3‑
酮为前体,通过与实施例1
‑3‑
1相同的方法(方法a)合成化合物19(橙色固体)。图15显示了1h nmr谱。
[0359]
产率47%;1h nmr(400mhz,dmso
‑
d6):δ10.19(s,2h),10.15(s,1h),9.80(s,1h),8.60(d,j=1.7hz,1h),8.43(dd,j=2.0,8.3hz,1h),7.57(d,j=8.3hz,1h),6.71
‑
6.67(m,4h),6.57(dd,j=2.2,8.7hz,2h);
13
c nmr(100mhz,cdcl3):δ185.0,167.5,159.7,152.6,147.7,137.4,129.2,128.0,127.9,126.8,126.0,116.7,112.7,108.9,102.3,83.6;esi
‑
tof ms(正模式)计算c
23
h
14
n3o6[m+h]
+
428.09m/z,求得428.10。
[0360]7‑
3.聚乙二醇系留三唑
‑4‑
甲醛(20)的合成(mw.~4kda)
[0361][0362]
以分子量约4kda的聚乙二醇单甲醚(东京化成工业株式会社制)为起始原料,采用与实施例1
‑3‑
1相同的方法合成化合物20(棕色固体),参考已发表的报告(mb van eldijk,fcm smit,n.vermue,mf debets,s.schoffelen,jcm van hest,biomacromolecules,2014,15,2751
‑
2759)。通过用乙醚再沉淀进行纯化。图16显示了1h nmr谱。
[0363]
产率50%;1h nmr(400mhz,cdcl3):δ10.14(s,1h),8.41(s,1h),4.63(t,j=4.8hz,1h),3.91
‑
3.45(m,366h)。
[0364]7‑
4.1
‑
(2
‑
(2
‑
(2
‑
(2
‑
叠氮乙氧基)乙氧基)乙氧基)乙基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(21)的合成
[0365][0366]
使用与实施例1
‑3‑
1相同的方法(方法a),使用1
‑
叠氮基
‑2‑
(2
‑
(2
‑
(2
‑
叠氮基乙氧基)乙氧基)乙氧基)乙烷作为前体合成化合物21(黄色油状物)。图17显示了1h nmr谱。
[0367]
产率27%;1h nmr(400mhz,cdcl3):δ10.1(s,1h),8.41(s,1h),4.62(t,j=4.8hz,2h),3.90(t,j=4.8hz,2h),3.68(m,4h),3.65
‑
3.63(m,6h),3.38(t,j=5.0hz,2h);13c nmr(100mhz,cdcl3):δ185.4,148.0,126.9,70.7(two signal were merged),70.7(两个信号合并),70.2,69.0,50.8,50.7;esi
‑
tof ms(正模式)计算c
11
h
18
n6o4na[m+na]
+
321.1m/z,求得321.1。
[0368]7‑
5.化合物22的合成
[0369]
使用与实施例6相同的方法(方法f),使用n
‑
(3
‑
氨基丙酰基)
‑
5,6
‑
二氢
‑
11,12
‑
二脱氢二苯并[b,f]氮杂环辛四稀作为前体合成化合物22(黄色固体)。图18显示了1h nmr谱。
[0370]
产率75%;1h nmr(400mhz,cd3cn):δ9.89(s,1h),7.88(s,1h),7.62(d,j=7.2hz,1h),7.45
‑
7.43(m,4h),7.39
‑
7.32(m,2h),7.22(dd,j=1.4,7.4hz,1h),5.06(d,j=14hz,1h),4.45(m,2h),3.66(d,j=14hz,1h),2.90(td,j=6.0,17hz,1h),2.35(td,j=6.0,17hz,1h);esi
‑
tof ms正模式)计算c
11
h
18
n6o4na[m+na]
+
321.1m/z,求得321.1。
[0371]
实施例8.肽n端修饰1
[0372]8‑
1.试剂、溶剂等
[0373]
使用的超纯水是通过使用millipore integral 3纯化获得的。作为其他试剂和溶剂,可直接使用市售产品。
[0374]8‑
2.肽n端修饰
[0375]
该方法针对肽的n端。可以作为靶标的肽是其中n端氨基未修饰并且n端的第二个氨基酸残基是脯氨酸以外的氨基酸的肽。作为一个具体实例,血管紧张素i的n端修饰如下
所述。
[0376]
下面显示了血管紧张素i的氨基酸序列。
[0377]
drvyihpfhl(seq id no:1)
[0378]
参考已发表的报告(j.i.macdonald,h.k.munch,t.moore,m.b.francis,nat.chem.biol.2015,11,326
‑
331)进行肽的n端修饰。下面描述具体的实验过程。
[0379]
化合物7在二甲基亚砜(dmso)中的溶液(200mm,1μl,0.2μmol,终浓度:10mm)用磷酸钾缓冲液(10mm,ph 7.5,17μl)稀释。向其中加入肽水溶液(1mm、2μl、2nmol、终浓度:100μm),并将混合物在37℃下振荡4小时。修饰百分比(=修饰肽/(总肽量))通过使用lc/ms从质谱中的峰强度评估。图19显示了结果。该反应条件下的修饰百分比计算为89%。上述结果证实,根据本发明的化合物用作蛋白质修饰剂。
[0380]
实施例9.肽n端修饰2
[0381]
所使用的试剂、设备、溶剂等与实施例8类似。作为化合物24,使用根据已发表的报告(h.hagiwara,s.okada,chem.commun.,2016,52,815
‑
818)合成的化合物。
[0382]9‑
1.肽n端修饰
[0383]
使用模型肽作为底物进行该修饰反应中产物的结构分析。作为具体实例,进行以下方案中所示的反应。
[0384][0385]
将化合物24(44mg,0.4mmol)在二甲基甲酰胺(dmf)中的溶液(200μl)加入化合物23(11mg,0.04mmol)的磷酸钾缓冲液(10mm,ph 7.5,1.8ml)中,并将混合物在37℃下振荡16小时。减压蒸馏除去溶剂。所得粗产物通过硅胶柱层析(乙酸乙酯:乙腈:水=95:5:0至0:95:5)纯化,得到化合物25。
[0386]9‑
2.化合物25的1h nmr分析
[0387]
图20显示了化合物25在氘代乙腈中的1h nmr谱归属结果。
[0388]
5.7ppm处的两个峰归属于4
‑
咪唑烷酮环的2
‑
位特异的质子h
e
,证实该反应在肽的n端形成4
‑
咪唑烷酮环。这两个峰来自异构体,其中不对称点是4
‑
咪唑烷酮环的2
‑
位。
[0389]
实施例10.蛋白质n端修饰1
[0390]
10
‑
1.试剂、溶剂等
[0391]
来自牛胰腺的核糖核酸酶a(rnase)购自roche。使用的超纯水是通过使用millipore integral 3纯化获得的。作为其他试剂和溶剂,可直接使用市售产品。
[0392]
10
‑
2.蛋白质修饰
[0393]
该方法针对蛋白质的n端。可以成为靶标的蛋白质是n端氨基未修饰并且n端的第二个氨基酸残基是脯氨酸以外的氨基酸的蛋白质。作为一个具体例子,下面描述了来自牛胰腺的核糖核酸酶a(rnase)的n端修饰。
[0394]
以下为rnase的氨基酸序列(pdb:1fs3)。
[0395][0396]
参考已发表的报告(j.i.macdonald,h.k.munch,t.moore,m.b.francis,nat.chem.biol.2015,11,326
‑
331)进行蛋白质的n端修饰。下面描述具体的实验过程。
[0397]
化合物7在二甲基亚砜(dmso)中的溶液(200mm,2.5μl,0.5μmol,终浓度:10mm)用磷酸钾缓冲液(10mm,ph 7.5,45μl)稀释。向其中加入rnase的超纯水溶液(1mm、2.5μl、2.5nmol、终浓度:50μm),将混合物在37℃下振荡16小时。修饰百分比(=修饰的rnase/(总rnase量))是通过使用lc/ms从质谱中的峰强度评估的。图21显示了结果。该反应条件下的修饰百分比计算为88%。反应后的产物根据需要通过尺寸排阻色谱法纯化。
[0398]
实施例11.用功能分子修饰蛋白质的n端
[0399]
11
‑
1.试剂、溶剂等
[0400]
来自牛胰腺的核糖核酸酶a(rnase)购自roche。使用的超纯水是通过使用millipore integral 3纯化获得的。作为其他试剂和溶剂,可直接使用市售产品。
[0401]
11
‑
2.用生物素修饰蛋白质的n端
[0402]
以化合物18为修饰剂,通过与实施例10
‑
2相同的方法进行生物素对蛋白质n端的修饰。图22显示了产物的lc/ms分析结果。生物素部分的修饰百分比计算为79%。
[0403]
11
‑
3.用荧光染料修饰蛋白质n端
[0404]
使用化合物19作为修饰剂,通过与实施例10
‑
2中相同的方法用荧光染料(此处为荧光素分子)对蛋白质的n端进行修饰。图23显示了产物的lc/ms分析结果。计算出关于荧光染料部分的修饰百分比为73%。
[0405]
11
‑
4.用叠氮基修饰蛋白质的n端
[0406]
使用化合物21作为修饰剂,通过与实施例10
‑
2中相同的方法用叠氮基对蛋白质的n
‑
端进行修饰。图24显示了产物的lc/ms分析结果。叠氮基的修饰百分比计算为79%。
[0407]
11
‑
5.用功能分子修饰,从修饰蛋白质n端的叠氮基开始
[0408]
将实施例11
‑
4中制备的叠氮基修饰的rnase通过应变促进的炔
‑
叠氮环加成反应用荧光染料修饰。在反应中使用了化合物26,即具有二苯并环辛炔部分作为炔底物的荧光素。图25显示了方案和结果。
[0409]
将化合物26在二甲基亚砜(dmso)中的溶液(10mm,1μl,终浓度:100μm)添加到含有rnase_21(rnase_21浓度:120μm,终浓度:20μm,缓冲液浓度:10mm,ph 7.5,82.3μl)的磷酸盐缓冲液中,并将混合物在4℃下静置16小时。关于荧光染料部分的修饰百分比计算为55%。
[0410]
11
‑
6.用应变炔部分修饰蛋白质的n端
[0411]
使用化合物22作为修饰剂通过与实施例10
‑
2中相同的方法用应变炔部分对蛋白质的n端进行修饰。图26显示了产物的lc/ms分析结果。环辛炔部分的修饰百分比计算为40%。
[0412]
实施例12.合成固定有反应物的树脂,用于非均相反应
[0413]
通过化合物10和实施例6中所示胺前体之间的重排反应构建新的n端选择性改性剂可用作不使用铜催化剂的清洁反应。因此,图27显示了通过将各种甲醛前体固定在树脂
或固体材料上来简化反应后产生的副产物(苯胺衍生物)的去除并且直接使用反应后的溶液进行蛋白质n端修饰的方法。
[0414]
12
‑
1.使用的设备、试剂、溶剂等
[0415]
所用设备、试剂、溶剂同实施例1、8。所用氨甲基聚苯乙烯树脂购自东京化学工业株式会社。
[0416]
12
‑
2.固定有反应物的树脂的合成
[0417]
作为具体实例,图28显示了其上固定有化合物27的聚苯乙烯树脂的合成。根据图29所示的方案进行合成。
[0418]
将氨甲基聚苯乙烯树脂(氨甲基/ps树脂,200mg)分散在dmf(30ml)中后,通过振荡10分钟使树脂溶胀。将5
‑
叠氮基
‑2‑
硝基苯甲酸(208mg,1.0mmol)、1
‑
羟基苯并三唑一水合物(200mg,1.3mmol)和n,n'
‑
二异丙基碳二亚胺(156μl,1.0mmol)添加到树脂分散液中,并将混合物摇晃过夜。过滤反应后的树脂,依次用二甲基甲酰胺
‑
纯水
‑
氯仿和丙酮洗涤,减压干燥。随后,为了使未反应的氨基失活,所得树脂用乙酸酐(1ml)和氯仿(4ml)的混合溶液处理30分钟,用氯仿
‑
甲醇和丙酮洗涤,减压干燥得到叠氮基修饰的聚苯乙烯树脂(叠氮基/ps树脂)。
[0419]
将叠氮基/ps树脂(220mg)用dmf/水(6:1,7ml)的混合溶液溶胀后,将五水合硫酸铜(ii)(25mg,0.1mmol)、抗坏血酸钠(40mg,0.2mmol)和炔丙醛二乙缩醛(356μl,2.5mmol)加入树脂分散液中,并将混合物室温振荡12小时。过滤反应后的树脂,依次用二甲基甲酰胺
‑
纯水
‑
氯仿、甲醇和丙酮洗涤。随后,将树脂用12%氨水溶液(5ml)处理10分钟,过滤,并用纯水洗涤以去除树脂表面上残留的铜催化剂。最后将所得树脂用盐酸水溶液/四氢呋喃(1:1,5ml)混合溶液处理3次,每次2分钟,过滤,用纯水和丙酮洗涤脱保护缩醛保护基团,从而得到固定有化合物27的树脂(27/ps树脂)。
[0420]
12
‑
3.树脂的鉴别
[0421]
通过红外光谱确定了化合物27固定在其上的树脂(27/ps树脂)表面上的化学物种。图30显示了结果。在叠氮基修饰的树脂(叠氮基/ps树脂)中,在2115cm
‑1处观察到叠氮基的伸缩振动特征。在通过cuaac反应形成三唑环后的树脂(27/ps树脂)中,叠氮伸缩振动峰消失,新观察到醛基伸缩振动引起的1699cm
‑1处的吸收。上述结果支持通过cuaac反应用醛基进行的修饰,表明制备了27/ps树脂。
[0422]
12
‑
4.蛋白质n端修饰
[0423]
使用所制备的固定有反应物的树脂(27/ps树脂)进行n端修饰剂的制备和后续的蛋白质n端修饰。作为一个具体实例,核糖核酸酶a(rnase a)n端修饰如下所述。
[0424]
将27/ps树脂(5mg)添加到苄胺的二甲亚砜溶液(100mm,50μl,5μmol)中,并使用加热块在100℃下加热混合物90分钟。将所得混合物空冷至室温后,将上清液(10μl,1μmol)用磷酸盐缓冲液(10mm,ph 7.5,85μl)稀释。向其中加入rnase a溶液(1mm、5μl、5nmol),并将混合物在37℃下振荡16小时。反应后,使用lc/ms进行修饰评价。即使在使用固定有反应物的树脂(27/ps树脂)的情况下,也获得了与使用分离纯化的n端修饰剂的情况相当的修饰百分比(75%)。该结果证明了通过使用固定有反应物的树脂(27/ps树脂)的后续反应对蛋白质进行n端修饰的方法。图31显示了结果。
[0425]
12
‑
5.固定有反应物的树脂的合成方法的改进
[0426]
在实施例12
‑
3中,在通过蛋白质修饰获得的样品中,观察到似乎向蛋白质添加了氧的产物。这被认为是因为用于树脂合成的铜催化剂残留在树脂上。因此,在新的合成方案下制备了27/ps树脂,并将其应用于蛋白质修饰。具体而言,根据图32所示的方案制备27/ps树脂。
[0427]
12
‑5‑
1.5
‑
叠氮基
‑2‑
硝基苯甲酸(28)的合成
[0428][0429]
将亚硝酸钠(727mg,10.5mmol)的水溶液(6ml)添加到含有5
‑
氨基
‑2‑
硝基苯甲酸(1.60g,8.9mmol)的浓hcl/etoh/h2o溶剂混合物(2:1:2,总共43ml)中,并将混合物在0℃搅拌1小时。随后,分批加入叠氮化钠(868mg,13.4mmol),并将溶液在0℃下剧烈搅拌1小时,然后在室温下搅拌1小时。
[0430]
反应溶液用超纯水稀释,过滤,然后用超纯水(20ml
×
2)洗涤。减压干燥,得到化合物28(淡黄色固体)。图33显示了1h nmr谱。
[0431]
产率37%;1h nmr(400mhz,dmso
‑
d6):δ,8.1(d,j=9.4hz,3h),7.43
‑
7.41(m,2h);
13
c nmr(100mhz,dmso
‑
d6):δ,165.8,145.3,143.4,130.8,126.2,121.7,119.6;esi
‑
tof ms(正模式)计算c7h4nan4o4[m+na]
+
231.012m/z,求得231.011。
[0432]
12
‑5‑
2.2,5
‑
二氧代吡咯烷
‑1‑
基
‑5‑
(4
‑
甲酰基
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)
‑2‑
硝基苯甲酸酯(30)的合成
[0433][0434]
将化合物28(1.75g,8.4mmol)和3,3
‑
二乙氧基丙
‑1‑
炔(1.2ml,1.08g,8.4mmol)分散在纯水(21ml)和叔丁醇(21ml)的混合物中。加入五水合硫酸铜(ii)(419mg,1.68mmol,20mol%)和抗坏血酸钠(666mg,3.36mmol,40mol%),在70℃、氮气氛下搅拌过夜。将反应液冷却至室温后,加入饱和食盐水停止反应,然后用乙酸乙酯(20ml
×
3)萃取。有机层用硫酸钠干燥,减压蒸馏除去溶剂,得到含有化合物29的粗品,省略纯化操作,将粗品直接用于下一步反应。
[0435]
将n
‑
羟基琥珀酰亚胺(764mg,6.64mmol)加入到含有粗产物的dmf溶液(22ml)中,并将混合物在室温下搅拌10分钟。随后,加入n,n'
‑
二环己基碳二亚胺(1.14g,5.5mmol),并将反应混合物搅拌过夜。滤出沉淀,减压干燥滤液以除去溶剂。将残余物溶解在乙酸乙酯(50ml)中,并再次滤出所得沉淀物。所得滤液用饱和食盐水(20ml
×
2)洗涤,有机层用硫酸钠干燥,并减压蒸馏除去溶剂,得到含有化合物29的粗产物。硅胶柱层析纯化得到化合物30(浅黄色固体)。图34显示了1h nmr谱。
[0436]
产率18%(两步后);1h nmr(400mhz,dmso
‑
d6):δ,10.15(s,1h),9.91(s,1h),8.64
‑
8.61(m,2h),8.57
‑
8.55(m,1h),2.91(s,4h);
13
c nmr(100mhz,dmso
‑
d6):δ,184.9,169.7,160.1,147.8,146.8,139.3,127.5,127.4,126.0,123.0,121.9,25.6;esi
‑
tof ms(正模式)计算c
14
h9nan5o7[m+na]
+
382.038)m/z,求得382.039。
[0437]
12
‑5‑
3.27/ps树脂合成2
[0438]
将氨甲基聚苯乙烯树脂(氨甲基/ps树脂,200mg)分散在dmf(5ml)中后,通过振荡1小时使树脂溶胀。将化合物30(150mg,0.42mmol)和n,n'
‑
二异丙基碳二亚胺(146μl,0.84mmol)加入到树脂的分散液中,并将混合物振荡过夜。过滤反应后的树脂,依次用二甲基甲酰胺
‑
纯水
‑
氯仿和丙酮洗涤,减压干燥。随后,为了使未反应的氨基失活,所得树脂用乙酸酐(1ml)和氯仿(4ml)的混合溶液处理30分钟,用氯仿
‑
甲醇和丙酮洗涤,减压干燥得到27/ps树脂。
[0439]
12
‑
6.蛋白质n端修饰
[0440]
使用固定有反应物的新制备的树脂(27/ps树脂)进行n端修饰剂的制备和后续的蛋白质n端修饰。作为一个具体实例,核糖核酸酶a(rnase a)n端修饰如下所述。图35显示了该方案。
[0441]
将27/ps树脂(5mg)添加到苄胺的二甲亚砜溶液(100mm,40μl,4μmol)中,并使用加热块在100℃下加热混合物90分钟。将所得混合物空冷至室温后,上清液(10μl,1μmol)用磷酸盐缓冲液(10mm,ph 7.5,85μl)稀释。向其中加入rnase a溶液(1mm、5μl、5nmol),并将混合物在37℃下振荡16小时。反应后,使用lc/ms进行修饰评价。图36显示了结果。即使在使用固定有反应物的树脂(27/ps树脂)的情况下,也获得了与使用分离纯化的n端修饰剂的情况相当的修饰百分比(82%)。此外,对于各种功能性胺化合物,证明了通过相同的方法可以对蛋白质n端进行特异性修饰。
[0442]
实施例13.在均相体系中利用迪姆罗特重排反应合成蛋白质修饰剂和后续的蛋白质修饰
[0443]
通过化合物10和实施例6中所示胺前体之间的重排反应构建新的n端选择性修饰剂可用作不使用铜催化剂的清洁反应。实施例12显示了非均相系统中的迪姆罗特重排反应以简化副产物的去除。另一方面,也推测存在作为副产物的苯胺衍生物的存在不会对蛋白质修饰造成问题的情况。因此,本部分显示了在均相系统中的迪姆罗特重排反应中合成甲醛衍生物和后续的蛋白质修饰。图37显示了该方案。
[0444]
13
‑
1.试剂、溶剂等
[0445]
所用的设备、试剂和溶剂与实施例1和8中的那些相似。来自牛胰腺的核糖核酸酶a(rnase)购自roche。使用的超纯水通过millipore integral 3纯化获得。
[0446]
13
‑
2.迪姆罗特重排反应条件的优化
[0447]
图38显示了迪姆罗特重排反应的反应机理。由于该反应包括亚胺形成作为第一步,因此可以预期加入酸催化剂以提高反应效率。因此,在各种酸催化剂的存在下,使用苄胺作为模型底物进行与化合物10的迪姆罗特重排反应。
[0448]
将化合物10在二甲基亚砜中的溶液(200mm、20μl、4μmol)和酸性水溶液(200mm或400mm、1μl、5mol%或10mol%)加入到苄胺在二甲基亚砜中的溶液中(200mm,20μl,4μmol),使用加热块在100℃下加热混合物30分钟。空冷至室温后,取1μl反应液用磷酸盐缓冲液
(100mm,ph 7.0,200μl)稀释,并转移至96孔板中,进行紫外
‑
可见吸收测定。反应的转化率是根据基于380nm处的吸收强度绘制的标准曲线计算的,这是对硝基苯胺(一种产物)的特征。图39显示了所用酸催化剂的结构,表1显示了获得的转化率。
[0449]
在表1中,转化率
a
是根据副产物对硝基苯胺的吸收强度计算的。
b
mops的添加量设定为10mol%。
[0450]
表1
[0451][0452]
上述结果表明,加入酸催化剂提高了转化率。建议加入磺酸是特别有效的。此外,还发现各种已知用作古德缓冲液的磺酸盐可以提高反应效率。一般来说,由于这些磺酸盐对蛋白质的功能或结构没有显著影响,因此迪姆罗特重排反应的混合溶液可直接用于后续的蛋白质修饰反应。选择3
‑
吗啉代丙磺酸(mops)作为酸催化剂,进行后续实验。
[0453]
13
‑
3.蛋白质n端修饰
[0454]
将化合物10在二甲基亚砜中的溶液(200mm、20μl、4μmol)和mops水溶液(200mm或400mm、1μl、5mol%或10mol%)添加到苄胺在二甲基亚砜中的溶液(200mm,20μl,4μmol)中,使用加热块在100℃下加热混合物30分钟。空冷至室温后,将反应混合物(5μl)用磷酸盐缓冲液(10mm,ph 7.5,42.5μl)稀释。向其中加入rnase a溶液(1mm,2.5μl,5nmol),并将混合物在37℃下振荡16小时。反应后,使用lc
‑
ms进行修饰评价。图40显示了lc
‑
ms分析的结果。
[0455]
使用各种功能性胺前体(例如炔烃、叠氮化物和荧光染料)制备蛋白质修饰剂,并实现了引入蛋白质的n端。此外,即使连续进行n端修饰剂的制备和蛋白质修饰反应,修饰百分比也与使用分离纯化的n端修饰剂的情况相当。结果证明了通过在均相系统中使用迪姆罗特重排反应的连续反应对蛋白质n端进行修饰的方法。
[0456]
实施例14.使用bis
‑
ta4c分子修饰蛋白质n端并通过肟形成引入功能分子
[0457]
在使用两末端具有氨基的二胺作为前体的迪姆罗特重排中,获得在两末端引入了具有ta4c部分的分子(以下称为“bis
‑
ta4c”)。在使用这种bis
‑
ta4c的蛋白质n端修饰中,可以将醛部分引入蛋白质的n端。从醛开始的化学修饰反应的一个例子包括与羟胺形成肟。该反应也适用于蛋白质修饰,因为它甚至在温和条件下的水中也能进行。此外,由于肟键是动态键,因此也可以根据需要去除功能性分子。因此,通过使用各种二胺作为前体,通过使用化合物10进行迪姆罗特重排反应来进行蛋白质修饰反应。
[0458]
14
‑
1.试剂、溶剂等
[0459]
所用的设备、试剂和溶剂与实施例1和8中的那些相似。来自牛胰腺的核糖核酸酶a(rnase)购自roche。使用的超纯水通过millipore integral 3纯化获得。
[0460]
14
‑
2.bis
‑
ta4c的制备及蛋白质修饰反应
[0461]
将化合物10在二甲亚砜中的溶液(200mm、20μl、4μmol)和酸性水溶液(200mm或400mm、1μl、5mol%或10mol%)添加到二胺31
‑
36的每种在二甲基亚砜(100mm,20μl,2μmol)的单独溶液中,并且使用加热块在90℃下单独加热混合物60分钟。空冷至室温后,将每个反应混合物(5μl)用磷酸盐缓冲液(10mm,ph 7.5,42.5μl)稀释。向其中加入rnase a溶液(1mm、2.5μl、5nmol),随后在37℃下振荡16小时。反应后,使用lc
‑
ms进行修饰评价。图41显示了方案,图42显示了蛋白质修饰的结果。
[0462]
当使用具有烷基链或低聚乙二醇链作为连接体的bis
‑
ta4c时,获得了良好的修饰蛋白(60%至82%)。相比之下,含有苯胺等芳香族骨架的bis
‑
ta4c不进行修饰反应。这被认为是由于bis
‑
ta4c的溶解度非常低。因此,引入提高水溶性的部分有望提高反应产率。
[0463]
14
‑
3.通过肟的形成将功能分子引入用bis
‑
ta4c修饰的蛋白质中
[0464]
从引入到具有bis
‑
ta4c的蛋白质n端的醛部分开始进行肟形成反应。具体而言,反应根据图43中所示的程序进行,参考已发表的报告(m.rashidian,mm mahmoodi,r.shah,jk dozier,cr wagner,md distefano,bioconjugate chem.2013,24,333
‑
342)。作为例子,下面描述了使用荧光染料和聚乙二醇的修饰。
[0465]
使用化合物35作为前体制备的bis
‑
ta4c修饰的rnase a(60μm,8.3μl,0.5nmol)用磷酸盐缓冲液(50mm,ph 7.0,35.7μl)稀释,并将含有羟胺37的二甲亚砜溶液(5mm、1μl、5nmol)和作为催化剂的间苯二胺(m
‑
pda)水溶液(50mm、5μl、0.25μmol)加入其中,随后在4℃下振荡6小时。反应后,使用lc
‑
ms进行修饰评价。图44显示蛋白质修饰的结果。尽管观察到一些作为催化剂的m
‑
pda的加合物,但证实以良好的产率实现了向肟加合物的转化。
[0466]
将使用化合物35作为前体制备的bis
‑
ta4c修饰的rnase a(60μm,8.3μl,0.5nmol)用磷酸盐缓冲液(50mm,ph 7.0,35.7μl)稀释;并向其中加入羟胺38(2mg,500nmol)和作为催化剂的间苯二胺(m
‑
pda)水溶液(50mm,5μl,0.25μmol),随后在室温下振荡16小时。反应后,使用sds
‑
page进行修饰评价。图45显示蛋白质修饰的结果。
[0467]
作为比较,还使用未修饰的蛋白质或具有羟基的聚乙二醇作为底物进行实验。仅在蛋白质n端的醛基和聚乙二醇中的羟胺部分的组合中观察到sds
‑
page上的条带偏移,证实了通过肟形成引入了聚乙二醇。
[0468]
实施例15.温和条件下的迪姆罗特重排反应
[0469]
在实施例13和14所示的均相体系中通过迪姆罗特重排反应制备n端修饰剂需要90℃至100℃的高反应温度。因此,需要进一步改进前体结构和反应条件以适应具有低耐热性的底物。为了在较低温度下实现反应,对作为前体的化合物10的结构进行了修饰。特别是,其中硝基苯基部分被改变为具有更高吸电子性的取代基的化合物可预期用作有用的前体,因为中间体稳定,并且逆反应不太可能进行。本节描述了根据该策略合成化合物以及应用于迪姆罗特重排反应的示例。
[0470]
15
‑
1.化合物合成
[0471]
作为具体的结构实例,具有4
‑
氰基四氟苯基的三唑甲醛40的合成描述如下。
[0472][0473]
15
‑1‑
1.4
‑
(4
‑
(二乙氧基甲基)
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)
‑
2,3,5,6
‑
四氟苄腈(39)的合成
[0474]
将含有五氟苄腈(850mg,541μl,4.4mmol)和叠氮化钠(260mg,4.0mmol)的乙腈溶液(10ml)在氮气气氛下在60℃搅拌16小时。随后,在氮气氛下加入炔丙醛二乙缩醛(570μl,4.0mmol)和碘化铜(i)(76mg,0.4mmol),并将混合物在室温下搅拌16小时。将反应溶液空冷至室温,然后用饱和氯化钠水溶液(40ml)稀释,接着用乙酸乙酯(50ml
×
3)萃取。将得到的有机层用硫酸镁干燥,将滤出固体的滤液减压蒸馏,得到粗产物。粗产物经硅胶柱层析纯化得到化合物39(白色固体)。图46显示了1h nmr谱。
[0475]
产率86%;1h
‑
nmr(400mhz,cdcl3):δ7.97(s,1h),5.82(s,1h),3.79
‑
3.64(m,4h),1.28(t,j=7.1hz,6h)。
[0476]
15
‑1‑
12.2,3,5,6
‑
四氟
‑4‑
(4
‑
甲酰基
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)苄腈(40)的合成
[0477]
将化合物39(688mg,2.0mmol)溶解在氯仿(4ml)中。向其中加入三氟乙酸(2ml),并将混合物在室温下搅拌16小时。减压蒸馏除去溶剂和三氟乙酸,得到粗产物。通过再沉淀(己烷:氯仿)纯化粗产物以得到化合物40(白色固体)。图47显示了1h nmr谱。
[0478]
产率78%;1h
‑
nmr(400mhz,cdcl3):δ10.26(s,1h),8.50(s,1h)。
[0479]
15
‑
2.以化合物40为前体的迪姆罗特重排反应
[0480]
为了评估化合物40的反应性,使用苄胺作为底物进行迪姆罗特重排反应。
[0481]
将化合物40的二甲亚砜溶液(100mm,40μl,4μmol)和mops水溶液(200mm,2μl,10mol%)加入苄胺的二甲亚砜溶液(100mm,40μl,4μmol)中,并使用加热块在40℃下加热混合物12小时。空冷至室温后,反应混合物用氘代二甲基亚砜(310μl)稀释。作为内标,加入1,3,5
‑
三甲氧基苯的氘代二甲基亚砜溶液(400mm,10μl,4mol),并进行1h nmr测量。产率由作为产物的化合物7的苄基位置的质子对应的峰的积分值基于内标的积分值算出。图48表示方案,图49示出结果。
[0482]
结果表明,与使用化合物10的情况相比,当使用化合物40时,迪姆罗特重排反应在低温下以良好的产率进行。这表明在三唑环的n1位置引入吸电子取代基对提高迪姆罗特重排反应的反应性有显著贡献。
[0483]
实施例16.蛋白质n端修饰2
[0484]
对通常用作蛋白质底物的人血清白蛋白(hsa)进行了n端修饰反应。
[0485]
16
‑
1.试剂、溶剂等
[0486]
人血清白蛋白(hsa)购自默克公司。使用的超纯水是通过使用millipore integral 3纯化获得的。作为其他试剂和溶剂,可直接使用市售产品。作为hsa,使用与已经在活体中经历过修饰的hsa的混合物。已发表的报告已确定修饰的hsa(a.kawakami,k.kubota,n.yamada,u.tagami,k.takehana,i.sonaka,e.suzuki,k.hirayama,febs j.,2006,273,3346
‑
3357。)。
[0487]
16
‑
2蛋白质修饰
[0488]
该方法针对蛋白质的n端。可以成为靶标的蛋白质是n端氨基未修饰的蛋白质且n端的第二个氨基酸残基是脯氨酸以外的氨基酸。作为具体实例,下文描述了人血清衍生白蛋白(hsa)的n端修饰。
[0489]
以下为hsa的氨基酸序列(pdb:1ao6)。
[0490][0491]
以化合物7为修饰剂,hsa为靶蛋白,采用与实施例10
‑
2相同的方法进行蛋白质n端修饰。图50显示了产物的lc/ms分析结果。修饰的蛋白质被称为“hsa1”。该反应条件下的修饰百分比计算为99%或更高。反应后的产物根据需要通过尺寸排阻色谱法纯化。
[0492]
实施例17.靶向蛋白质n端和半胱氨酸残基的蛋白质双重修饰
[0493]
17
‑
1.试剂、溶剂等
[0494]
所用设备、试剂、溶剂等与实施例16类似。
[0495]
17
‑
2.蛋白质中的半胱氨酸残基修饰
[0496]
参考已发表的报告(x.chen,h.wu,c.
‑
m.park,t.h.poole,g.keceli,n.o.devarie
‑
baez,a.w.tsang,w.t.lowther,l.b.poole,s.b.king,m.xian,c.m.furdui,acs chem.biol.,2017,12,2201
‑
2208)进行蛋白质中的半胱氨酸残基修饰。下面描述具体的实验过程。
[0497]
将化合物41在dmso/水(1:1)混合物中的溶液(25mm,40μl,1μmol,终浓度:500μm)用磷酸盐缓冲液(100mm,ph 7.0,1.86ml)稀释。向其中加入hsa的超纯水溶液(1mm、100μl、100nmol、终浓度:50μm),并将混合物在4℃下静置12小时。反应后,使用lc/ms进行修饰评价。图51显示了结果。修饰的蛋白质被称为“hsa2”。该反应条件下的修饰百分比计算为99%或更高。反应后的产物根据需要通过尺寸排阻色谱法纯化。
[0498]
17
‑
3.其中半胱氨酸残基被修饰的蛋白质的n端修饰
[0499]
使用化合物7对实施例17
‑
2中制备的hsa2进行蛋白质n端的修饰。图52显示了方案和结果。
[0500]
将化合物7在二甲亚砜(dmso)中的溶液(200mm,1μl,终浓度:10mm)加入到含有hsa2的磷酸盐缓冲溶液(hsa2浓度:200μm,终浓度:50μm,缓冲溶液浓度:10mm,ph 7.5,19μl)中,并将混合物在37℃下振荡16小时。修饰的蛋白质被称为“hsa3”。该反应条件下的修饰百分比计算为72%。
[0501]
实施例18.在n端修饰的蛋白质的稳定性
[0502]
18
‑
1.试剂、溶剂等
[0503]
所用设备、试剂、溶剂等与实施例10类似。
[0504]
18
‑
2.修饰蛋白随时间的稳定性
[0505]
将含有rnase_7的磷酸盐缓冲液(rnase_7浓度:100μm,终浓度:10μm,缓冲溶液浓度:100mm,ph 7.0,10μl)用磷酸盐缓冲液(100ml,ph 7.0,90μl)稀释,并在37℃下静置12小时、24小时和48小时。例如,图53显示了静置24小时后的lc/ms分析结果。
[0506]
涉及在n端形成4
‑
咪唑啉酮环的该修饰反应是平衡反应。从lc/ms分析的结果可以认为,未修饰的蛋白质和修饰剂通过在n端修饰的蛋白质中的4
‑
咪唑烷酮环水解(其是逆反应)而再生。图54显示了一张图表,其纵轴表示由质谱中的峰的强度而计算的释放量(=1
‑
(静置后的修饰rnase/总rnase量)/(静置前的修饰rnase/总rnase量));横轴表示rnase静置的时间。
[0507]
18
‑
3.修饰蛋白随ph变化的稳定性
[0508]
将含有rnase_7的磷酸盐缓冲液(rnase_7浓度:100μm,终浓度:10μm,缓冲溶液浓度:100mm,ph 7.0,10μl)用缓冲液(100ml,90μl)稀释,使ph为4、5、6、7、8,37℃放置12小时。对于ph 4和5的条件使用醋酸盐缓冲液进行稀释,对于ph 6、7和8的条件使用磷酸盐缓冲液进行稀释。图55显示了释放的结果。
[0509]
实施例19.化合物合成8(1
‑
和5
‑
位具有取代基的三唑甲醛的合成)
[0510]
合成中使用的试剂和溶剂直接使用市售品。作为前体的叠氮化合物、炔化合物和硼酸频哪醇酯是参考已发表的报道(l.s.c.
‑
verduyn,l.mirfeizi,r.a.dierckx,p.h.elsinga,b.l.feringa,chem.commun.,2009,16,2139
‑
2140;e.jahnke,j.weiss,s.neuhaus,t.n.hoheisel h.frauenrath,chem.eur.j.,2009,15,388
‑
404;j.c.pieck,d.kuch,f.grolle,u.linne,c.haas,t.carell,j.am.chem.soc.,2006,128,1404
‑
1405;j.schmidt,m.rotter,t.weiser,s.wittmann,l.weizel,a.kaiser,j.heering,t.goebel,c.angioni,m.wurglics,a.paulke,g.geisslinger,a.kahnt,d.steinhilber,e.proschak,d.merk,j.med.chem.,2017,60,7703
‑
7724;和jr.white,g.j.price,s.schiffers,p.r.raithby,p.k.plucinski,c.g.frost,tetrahedron letters,2010,51,3913
‑
3917)合成的。
[0511]
19
‑
1.1
‑
苄基
‑5‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛的合成(44)
[0512]
化合物44是根据以下方案合成的,参考已发表的报道(k.yamamoto,t.bruun,j.y.kim,l.zhang,m.lautens,org.lett.,2016,18,2644
‑
2647;和j.deng,y.
‑
m.wu,q.
‑
y.chen,synthesis,2005,16,2730
‑
2738).
[0513][0514]
19
‑1‑
1.1
‑
苄基
‑5‑
碘
‑
1h
‑
1,2,3
‑
三唑
‑4‑
基)甲醇(42)的合成
[0515]
在氮气氛下将thf(89ml)加入苄基叠氮化物(0.67g,5.0mmol)、3
‑
碘丙
‑2‑
炔基
‑1‑
醇(0.91g,5.0mmol)、碘化铜(i)(95mg,0.50mmol)、tbta(0.27g,0.50mmol)和乙酸钾(1.5g,15mmol)的混合物中,并将混合物在室温下搅拌过夜。减压蒸馏除去反应混合物的溶剂,用水(30ml)和乙酸乙酯(30ml)稀释,然后用乙酸乙酯(30ml
×
3)萃取。将所得有机层用饱和盐水(30ml
×
2)洗涤并用硫酸钠干燥,减压蒸馏除去过滤所得滤液的溶剂。残余物通过硅胶柱层析(己烷:乙酸乙酯)纯化,得到化合物42(白色固体)。图56显示了1h nmr谱。
[0516]
产率82%:1h nmr(400mhz,dmso
‑
d6):δ,7.42
‑
7.33(m,3h),7.24
‑
7.22(m,2h),5.65(s,2h),5.27(t,j=5.6hz,1h),4.47(d,j=5.6hz,2h):
13
c nmr(100mhz,dmso
‑
d6):δ,151.30,135.52,128.89,128.20,127.54,83.79,54.99,53.25:esi
‑
tof ms(正模式)计算c
10
h
10
in3nao[m+na]
+
337.976m/z,求得337.977。
[0517]
19
‑1‑
2.(1
‑
苄基
‑5‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
基)甲醇(43)的合成
[0518]
将纯水(2.4ml)加入化合物42(0.24g,0.75mmol)、苯基硼酸(0.18g,1.5mmol)、碳酸钾(0.21g,1.5mmol)和乙酸钯(17mg,0.075毫摩尔)的混合物中。此后,在氮气氛下加入thf(9.6ml),并将混合物在70℃下搅拌过夜。在减压下蒸馏除去反应混合物的溶剂,用水(30ml)和乙酸乙酯(30ml)进行稀释,然后用乙酸乙酯(30ml
×
3)萃取。将所得有机层用饱和盐水(30ml
×
2)洗涤并用硫酸钠干燥,并在减压下蒸馏出过滤后所得滤液的溶剂。残余物通过快速柱色谱法(己烷:乙酸乙酯)纯化,得到化合物43(浅黄色固体)。图57显示了1h nmr谱。
[0519]
产率54%;1h nmr(400mhz,cdcl3):δ,7.49
‑
7.42(m,3h),7.29
‑
7.23(m,5h),7.06
‑
7.04(m,2h),5.48(s,2h),4.68(d,j=6.0hz,2h),2.16(t,j=6.0hz,1h):esi
‑
tof ms(正模式)计算c
16
h
15
n3nao[m+na]
+
288.111m/z,求得288.112。
[0520]
19
‑1‑
3.1
‑
苄基
‑5‑
苯基
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(44)的合成
[0521]
将活化的二氧化锰(163mg,1.9mmol)加入到化合物43(50mg,0.19mmol)在1,4
‑
二恶烷中的溶液(10ml)中,并将混合物在室温下搅拌过夜。过滤反应混合物,减压蒸馏除去所得滤液的溶剂。此后,所得粗产物通过快速柱色谱法(己烷:乙酸乙酯)纯化,得到化合物44(油)。图58显示了1h nmr谱。
[0522]
产率91%;1h nmr(400mhz,cdcl3):δ,10.14(s,1h),7.56
‑
7.46(m,3h),7.31
‑
7.27(m,5h),7.07
‑
7.05(m,2h),5.49(s,2h):esi
‑
tof ms(正模式)计算c
16
h
13
nan3o[m+na]
+
286.095m/z,求得286.093.
[0523]
19
‑1‑
4.(1
‑
苄基
‑5‑
(4
‑
甲氧基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
基)甲醇(45)的合成
[0524][0525]
以4
‑
甲氧基苯基硼酸为前体,通过与实施例19
‑1‑
2相同的方法合成化合物45(淡黄色固体)。图59显示了1h nmr谱。
[0526]
产率97%;1h nmr(400mhz,cdcl3):δ,7.29
‑
7.27(m,3h),7.17(d,j=8.8hz,2h),7.08
‑
7.07(m,2h),6.95(d,j=8.8hz,2h),5.46(s,2h),4.67(d,j=6.0hz,2h),3.85(s,3h),2.09(t,j=6.0hz,1h):
13
c nmr(100mhz,cdcl3):δ,160.75,144.90,135.95,135.63,131.16,128.95,128.32,127.43,118.45,114.62,56.09,55.53,52.07:esi
‑
tof ms(正模式)计算c
17
h
17
n3nao2[m+na]
+
318.121m/z,求得318.121.
[0527]
19
‑1‑
5.1
‑
苄基
‑5‑
(4
‑
甲氧基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(46)的合成
[0528][0529]
以化合物45为前体,通过与实施例19
‑1‑
3相同的方法合成化合物46(油)。图60显示了1h nmr谱。
[0530]
产率91%:1h nmr(400mhz,cdcl3):δ,10.14(s,1h),7.32
‑
7.30(m,3h),7.23(d,j=8.8hz,2h),7.10
‑
7.08(m,2h),6.99(d,j=8.8hz,2h),5.49(s,2h),3.87(s,3h):
13
c nmr(100mhz,cdcl3):δ,184.68,161.42,143.49,140.76,134.72,131.23,129.04,128.59,127.44,116.48,114.54,55.49,51.85:esi
‑
tof ms(正模式)计算c
17
h
15
n3nao2[m+na]
+
316.106m/z,求得316.104。
[0531]
19
‑1‑
6.(1
‑
苄基
‑5‑
(4
‑
硝基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
基)甲醇(47)的合成
[0532][0533]
通过与实施例19
‑1‑
2中相同的方法使用4
‑
硝基苯基硼酸作为前体合成化合物47(棕色固体)。图61显示了1h nmr谱。
[0534]
产率58%:1h nmr(400mhz,cdcl3):δ,8.28(d,j=8.8hz,2h),7.46(d,j=8.8hz,2h),7.31
‑
7.29(m,3h),7.05
‑
7.03(m,2h),5.52(s,2h),4.69(d,j=6.0hz,2h),2.14(t,j=6.0hz,1h):
13
c nmr(100mhz,cdcl3):δ,148.56,145.90,134.85,134.12,133.18,130.84,129.22,128.77,127.25,124.22,55.83,52.74:esi
‑
tof ms(正模式)计算c
16
h
14
n4nao3[m+na]
+
333.096m/z,求得333.095。
[0535]
19
‑1‑
7.1
‑
苄基
‑5‑
(4
‑
硝基苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(48)的合成
[0536][0537]
以化合物47为前体,通过与实施例19
‑1‑
3相同的方法合成化合物48(黄色固体)。图62显示了1h nmr谱。
[0538]
产率93%:1h nmr(400mhz,cdcl3):δ,10.20(s,1h),8.31(d,j=8.8hz,2h),7.43(d,j=8.8hz,2h),7.33
‑
7.28(m,3h),7.04
‑
7.02(m,2h),5.52(s,2h):
13
c nmr(100mhz,cdcl3):δ,185.02,149.11,144.27,137.67,133.99,131.05,129.37,129.15,127.45,124.07,52.64:esi
‑
tof ms(正模式)计算c
16
h
12
n4nao3[m+na]
+
331.080m/z,求得331.082。
[0539]
19
‑1‑
8.1
‑
(4
‑
((4
‑
(羟甲基)
‑5‑
碘
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)甲基)苯基)乙烷
‑1‑
酮(49)的合成
[0540][0541]
使用1
‑
(4
‑
(叠氮甲基)苯基)乙烷
‑1‑
酮作为前体,通过与实施例19
‑1‑
1相同的方法合成化合物49(白色固体)。图63显示了1h nmr谱。
[0542]
产率40%:1h nmr(400mhz,cdcl3):δ,7.94(d,j=8.3hz,2h),7.33(d,j=8.3hz,2h),5.65(s,2h),4.74(s,2h),2.59(s,3h),2.15(s,3h);
13
c nmr(100mhz,cdcl3):δ,197.5,151.5,139.1,137.4,129.1,128.1,78.9,56.8,53.9,26.8:esi
‑
tof ms(正模式)计算c
12
h
12
in3nao2[m+na]
+
379.987m/z,求得379.987。
[0543]
19
‑1‑
9.1
‑
(4
‑
((5
‑
(4
‑
(叠氮甲基)苯基)
‑4‑
(羟甲基)
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)甲基)苯基)乙烷
‑1‑
酮(50)的合成
[0544][0545]
使用化合物49和2
‑
(4
‑
(叠氮甲基)苯基)
‑
4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧杂硼烷作为前体,通过与实施例19
‑1‑
2相同的方法合成化合物50(淡黄色固体)。图64显示了1h nmr谱。
[0546]
产率71%:1h nmr(400mhz,cdcl3):δ,7.87(d,j=8.0hz,2h),7.40(d,j=8.0hz,2h),7.26(d,j=8.0hz,2h),7.13(d,j=8.0hz,2h),5.54(s,2h),4.69(d,j=5.8hz,2h),4.42(s,2h),2.57(s,3h),2.17(t,j=5.8hz,1h);13c nmr(100mhz,cdcl3):δ,197.5,145.4,140.3,137.5,137.1,135.6,130.2,129.1,128.9,127.5,126.3,55.9,54.4,51.9,26.8:esi
‑
tof ms(正模式)计算c
19
h
18
n6nao2[m+na]
+
385.138m/z,求得385.138。
[0547]
19
‑1‑
10.1
‑
(4
‑
乙酰苄基)
‑5‑
(4
‑
(叠氮甲基)苯基)
‑
1h
‑
1,2,3
‑
三唑
‑4‑
甲醛(51)的合成
[0548][0549]
以化合物50为前体,通过与实施例19
‑1‑
3相同的方法合成化合物51(澄清、油状)。图65显示了1h nmr谱。
[0550]
产率85%:1h nmr(400mhz,cdcl3):δ,10.17(s,1h),7.88(d,j=8.0hz,2h),7.44(d,j=8.0hz,2h),7.28(d,j=8.0hz,2h),7.14(d,j=8.0hz,2h),5.55(s,2h),4.45(s,2h),2.58(s,3h);13c nmr(100mhz,cdcl3):δ,197.4,184.7,143.9,140.0,139.3,138.6,137.4,130.1,129.1,128.7,127.7,124.6,54.3,51.7,26.8:esi
‑
tof ms(正模式)计算c
19
h
16
n6nao2[m+na]
+
383.123m/z,求得383.123。
[0551]
实施例20.肽n端修饰3
[0552]
参考已发表的报告(j.i.macdonald,h.k.munch,t.moore,m.b.francis,nat.chem.biol.2015,11,326
‑
331)对肽的n端进行修饰。下面描述具体的实验过程。
[0553]
将化合物44在二甲基亚砜(dmso)中的溶液(200mm,2μl,0.4μmol,终浓度:10mm)用磷酸盐缓冲液(10mm,ph 7.5,34μl)稀释。向其中加入肽水溶液(1mm、4μl、4nmol、终浓度:100μm),并将混合物在37℃下振荡16小时。修饰百分比(=(修饰肽量)/(总肽量))通过使用
lc/ms从质谱中的峰强度评估。图66显示了结果。该反应条件下的修饰百分比计算为>99%。
[0554]
实施例21.蛋白质n端修饰4
[0555]
参考已发表的报告(j.i.macdonald,h.k.munch,t.moore,m.b.francis,nat.chem.biol.2015,11,326
‑
331)进行蛋白质n端修饰。下面描述具体的实验过程。
[0556]
将化合物44在二甲基亚砜(dmso)中的溶液(200mm,5μl,1.0μmol,终浓度:10mm)用磷酸盐缓冲液(10mm,ph 7.5,90μl)稀释。向其中加入rnase的超纯水溶液(1mm、5μl、5nmol、终浓度:50μm),在37℃下振荡16小时。通过使用lc/ms从质谱中的峰强度评估修饰百分比(=(修饰的rnase)/(总rnase量))。图67显示了结果。该反应条件下的修饰百分比计算为48%。
[0557]
实施例22.用功能分子修饰蛋白质的n端
[0558]
22
‑
1.用乙酰基和叠氮基对蛋白质n端进行双重修饰
[0559]
使用化合物51作为修饰剂,通过与实施例21中相同的方法用乙酰基和叠氮基对蛋白质的n端进行双重修饰。图68显示了产物的lc/ms分析结果。乙酰基和叠氮基的修饰百分比计算为28%。
[0560]
22
‑
2.用功能分子修饰,从修饰蛋白质n端的叠氮基开始
[0561]
实施例22
‑
1中制备的叠氮基修饰的rnase通过应变促进的炔
‑
叠氮环加成反应用荧光染料修饰。在反应中使用了化合物26,即具有二苯并环辛炔部分作为炔底物的荧光素。图69显示了方案和结果。
[0562]
将化合物26在二甲基亚砜(dmso)中的溶液(8mm,1μl,终浓度:400μm)加入含有rnase_51的磷酸盐缓冲液(rnase_51浓度:76μm,终浓度:20μm,缓冲溶液浓度:10mm,ph 7.5,19μl)中,并将混合物在室温下静置6小时。关于荧光染料部分的修饰百分比计算为>99%。
[0563]
实施例23.将聚合物分子引入蛋白质的n端
[0564]
23
‑
1.具有三唑甲醛部分的聚乙二醇的合成
[0565]
合成了其中引入了三唑甲醛部分的聚乙二醇54。参考已发表的报道(l.rocard,a.berezin,f.de leo,d.bonifazi,angew.chem.int.ed.,2015,54,15739
‑
15743;and m.b.van eldijk,f.c.m.smit,n.vermue,m.f.debets,s.schoffelen,j.c.m.van hest,biomacromolecules,2014,15,2751
‑
2759),按照以下方案进行合成。
[0566][0567]
23
‑1‑
1.4
‑
((4
‑
甲酰基
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)甲基)苯甲酸(52)的合成
[0568]
通过使用与实施例1
‑3‑
1中相同的方法(方法a),使用(4
‑
叠氮甲基)苯甲酸作为前体合成化合物52(白色固体)。图70显示了1h nmr谱。
[0569]
产率71%;1h nmr(400mhz,dmso
‑
d6):δ,10.02(s,1h),9.00(s,1h),7.94(d,j=8.0hz,2h),7.43(d,j=8.0hz,2h),5.79(s,2h);
13
c nmr(100mhz,dmso
‑
d6):δ,185.0,166.9,147.1,140.1,130.7,129.9,128.7,128.2,52.8;esi
‑
tof ms(正模式)计算c
11
h9n3o3na[m+na]
+
254.054m/z,求得254.053。
[0570]
23
‑1‑
2.2,5
‑
二氧代吡咯烷
‑1‑
基
‑4‑
((4
‑
甲酰基
‑
1h
‑
1,2,3
‑
三唑
‑1‑
基)甲基)苯甲酸酯(53)的合成
[0571]
将n
‑
羟基琥珀酰亚胺(138mg,1.2mmol)和n,n'
‑
二环己基碳二亚胺(206mg,1.0mmol)加入到含有化合物52(231mg,1.0mmol)的thf溶液(10ml)中,在氮气气氛下于0℃搅拌该混合物24小时。滤出沉淀,滤液用乙酸乙酯(50ml)稀释,用饱和碳酸氢钠水溶液(20ml
×
2)和饱和食盐水(20ml
×
2)洗涤。有机层用硫酸钠干燥,减压蒸馏除去溶剂,得到粗产物。粗产物经硅胶柱层析纯化得到化合物53(白色固体)。图71显示了1h nmr谱。
[0572]
产率71%;1h nmr(400mhz,cdcl3):δ,10.15(s,1h),8.16(d,j=8.5hz,2h),8.07(s,1h),7.42(d,j=8.5hz,2h),5.70(s,2h),2.91(s,4h);
13
c nmr(100mhz,cdcl3):δ,185.0,169.1,161.3,148.4,140.5,131.7,128.5,126.3,125.4,54.1,25.8;esi
‑
tof ms(正模式)计算c
15
h
12
n4o5na[m+na]
+
351.070m/z,求得351.069。
[0573]
23
‑1‑
2.聚乙二醇系留三唑
‑4‑
甲醛(54)的合成
[0574]
将氨基封端的聚乙二醇(分子量:约4000、400mg、0.1mmol)和三乙胺(45l,0.32mmol)加入到含有化合物53(53mg,0.16mmol)的二氯甲烷(15ml)中,并将混合物在氮气氛下搅拌24小时。减压蒸馏除去溶剂后,残余物用硅胶柱色谱纯化。所得油状粗产物通过再沉淀(乙醚/二氯甲烷)纯化,得到化合物54(白色固体)。
[0575]
23
‑
2.使用化合物54对蛋白质n端进行修饰
[0576]
使用化合物54用聚乙二醇对蛋白质的n端进行修饰。以下是使用核糖核酸酶作为底物的实例。
[0577]
23
‑2‑
1.试剂、溶剂等
[0578]
来自牛胰腺的核糖核酸酶a(rnase)购自roche。使用的超纯水是通过使用millipore integral 3纯化获得的。作为其他试剂和溶剂,可直接使用市售产品。
[0579]
23
‑2‑
2.聚乙二醇修饰蛋白质n端
[0580]
以化合物54为修饰剂,通过与实施例10
‑
2相同的方法,用聚乙二醇对蛋白质的n端进行修饰。图72显示产物的sds
‑
page分析结果。聚乙二醇部分的修饰百分比计算为50%。
[0581]
23
‑2‑
3.用聚合物修饰蛋白质的n端,其中半胱氨酸残基被修饰
[0582]
通过与实施例17
‑
3相同的方法,使用化合物54作为修饰剂对hsa2蛋白的n端进行聚乙二醇修饰。反应得到的hsa(简称“hsa4”),其中半胱氨酸残基被叠氮基修饰,n端被聚乙二醇修饰,通过使用化合物26的应变促进炔
‑
叠氮环加成反应用荧光染料修饰。修饰的蛋白质称为“hsa5”。
[0583]
将化合物26的二甲基亚砜(dmso)溶液(6mm,1μl,终浓度:300μm)加入到含有hsa4的磷酸盐缓冲液(hsa4浓度:62μm,终浓度:10μm,缓冲液浓度:10mm,ph 7.5,19μl)中,并将混合物在4℃下静置6小时。对hsa和hsa2也进行相同的操作,并使用sds
‑
page分析产物。图73显示了方案和结果。
[0584]
当与化合物26反应时,仅在hsa2和hsa4的泳道(均用化合物41修饰以引入叠氮基团)中观察到荧光。此外,仅在化合物54修饰的hsa4泳道出现分子量增加的新条带,该条带是聚乙二醇衍生物。这些结果证实蛋白质中进行了聚乙二醇修饰和荧光染料修饰。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1