光学活性双膦基甲烷、其制造方法以及过渡金属络合物和不对称催化剂与流程
1.本发明涉及一种新颖的双膦基甲烷衍生物及其制造方法,以及过渡金属络合物、不对称催化剂和使用该催化剂制造有机化合物的方法。
背景技术:
2.在磷原子上具有不对称中心的光学活性膦配体在使用过渡金属络合物的催化不对称合成反应中起重要作用。作为在磷原子上具有不对称中心的光学活性膦配体,专利文献1中提出了1,2-双(二烷基膦基)苯衍生物。
3.在专利文献2中,提出了光学活性的2,3-双(二烷基膦基)吡嗪衍生物。该吡嗪衍生物具有源自吡嗪骨架的显著高的吸电子性,因此可以高效地进行将磷原子引入杂环的反应,该反应通常容易得到低产率。此外,由于吡嗪衍生物的磷原子具有电子密度低的特征,因此将具有吡嗪衍生物作为配体的金属络合物用作反应催化剂来充分利用该特征是有效的。
4.专利文献3和专利文献4提出了光学活性双膦基甲烷。具有光学活性双膦基甲烷作为配体的过渡金属络合物具有优异的不对称催化能力,但由于它以液体或油状物的形式提供,因此难以处理。此外,过渡金属络合物必须小心处理,因为膦配体在空气中容易被氧化。
5.引用列表
6.专利文献
7.专利文献1:日本专利特开第2000-319288号
8.专利文献2:日本专利特开第2007-56007号
9.专利文献3:日本专利特开第2000-136193号
10.专利文献4:wo2005/097370
技术实现要素:
11.技术问题
12.本发明的一个目的是提供一种新颖的光学活性双膦基甲烷,其适用作不对称催化剂的配体、在空气中的抗氧化性优异并且易于处理。目的还在于提供使用具有优异不对称催化能力的光学活性双膦基甲烷作为配体的过渡金属络合物。
13.问题解决方案
14.在对光学活性双膦基甲烷进行研究的过程中,本发明人发现一种新颖的光学活性双膦基甲烷,其在空气中不易氧化、因为在室温(25℃)下为固体而易于处理,且适用作不对称催化剂的配体,并且该发现导致本发明的完成。
15.第一发明是一种光学活性双膦基甲烷,其由以下通式(1)表示。
16.[式1]
[0017][0018]
其中r1表示金刚烷基;r2表示具有3个或更多个碳原子的支链烷基;并且*表示磷原子上的不对称中心。
[0019]
第二发明是用于制造第一发明的光学活性双膦基甲烷的方法,其包括:
[0020]
第一步:制备锂化膦硼烷,该锂化膦硼烷通过使由以下通式(2)表示的膦硼烷锂化而制得:
[0021]
[式2]
[0022][0023]
其中r1表示金刚烷基,和
[0024]
制备由以下通式(4)表示的光学活性膦硼烷衍生物,该光学活性膦硼烷衍生物通过使由以下通式(3)表示的光学活性羟甲基膦硼烷的羟基转化为离去官能团而制得:
[0025]
[式3]
[0026][0027]
其中r2表示具有3个或更多个碳原子的支链烷基;并且*表示磷原子上的不对称中心,
[0028]
[式4]
[0029][0030]
其中r2表示具有3个或更多个碳原子的支链烷基;a表示羟基的活化官能团;并且*表示磷原子上的不对称中心;
[0031]
第二步:使锂化膦硼烷与由通式(4)表示的光学活性膦硼烷衍生物反应,以获得光学活性双膦基甲烷硼烷;以及
[0032]
第三步:使光学活性双膦基甲烷硼烷脱硼。
[0033]
第三发明是一种过渡金属络合物,其包含第一发明的光学活性双膦基甲烷作为配体。
[0034]
第四发明是一种不对称催化剂,其包含第三发明的过渡金属络合物。
[0035]
发明的有益效果
[0036]
根据本发明,可以提供一种新颖的光学活性双膦基甲烷,其适用作不对称催化剂的配体、在空气中的抗氧化性优异且易于处理。此外,通过将使用本发明的光学活性双膦基甲烷作为配体的过渡金属络合物用作不对称催化剂,该不对称催化剂在不对称加氢反应中
表现出高对映选择性和高反应活性,并且目标物质可以以高光学纯度和高产率获得。
具体实施方式
[0037]
在下文中,将基于优选实施方式对本发明进行描述。
[0038]
在由通式(1)表示的本发明的光学活性双膦基甲烷中,r1是金刚烷基。
[0039]
在r1是金刚烷基的情况下,光学活性双膦基甲烷在室温下为固体,并且在使用其作为配体的过渡金属络合物用作不对称催化剂的情况下,表现出高对映选择性。
[0040]
通式(1)的r2表示具有3个或更多个碳原子的支链烷基。具有3个至更多个碳原子的支链烷基的实例包括具有3个至8个碳原子的支链烷基,比如异丙基、叔丁基和1,1,3,3-四甲基丁基(在一些情况下也称为“叔辛基”)。在本发明中,r2优选为叔丁基。
[0041]
在r2是具有3个或更多个碳原子的支链烷基的情况下,在使用光学活性双膦基甲烷作为配体的过渡金属络合物用作不对称催化剂的情况下,过渡金属络合物表现出高反应活性。
[0042]
尽管常规的光学活性双膦基甲烷在室温为液体或油状物,但本发明的光学活性双膦基甲烷在室温呈固体状态也是本发明的特征之一。因此,本发明的光学活性双膦基甲烷易于处理。此外,本发明的光学活性双膦基甲烷在空气中的抗氧化性优异也是特征之一。在此,在室温状态为固体是指在25℃的状态为固体。
[0043]
在下文中,对用于制造根据本发明的由通式(1)表示的光学活性双膦基甲烷的方法进行描述。
[0044]
本发明的制造方法包括:制备锂化膦硼烷和由通式(4)表示的光学活性膦硼烷衍生物,该锂化膦硼烷通过使由通式(2)表示的膦硼烷锂化而制得,该光学活性膦硼烷衍生物通过使由通式(3)表示的光学活性羟甲基膦硼烷的羟基转化为离去官能团而制得;使这些物质反应以获得光学活性双膦基甲烷硼烷;以及然后使光学活性双膦基甲烷硼烷脱硼。
[0045]
也就是说,本发明的用于制造由通式(1)表示的光学活性双膦基甲烷的方法包括以下三个步骤。
[0046]
(1)制备锂化膦硼烷和制备光学活性膦硼烷衍生物的第一步
[0047]
(2)获得光学活性双膦基甲烷硼烷的第二步
[0048]
(3)进行脱硼反应的第三步
[0049]
第一步是以下的步骤:制备锂化膦硼烷,该锂化膦硼烷通过使由通式(2)表示的膦硼烷锂化而制得,和制备由通式(4)表示的光学活性膦硼烷衍生物,该光学活性膦硼烷衍生物通过使由通式(3)表示的光学活性羟甲基膦硼烷的羟基转化为离去官能团而制得。此处,这些化合物的制备顺序没有特别限制。
[0050]
由通式(2)表示的膦硼烷可以通过熟知的方法制造。制造方法的实例包括日本专利特开第2001-253889号、第2003-300988号、第2007-70310号和第2010-138136号以及《有机化学杂志(j.org.chem.)》,2000,第65卷,第4185-4188页中所描述的方法。
[0051]
锂化膦硼烷的制备可以通过使由通式(2)表示的膦硼烷溶解在溶剂中,然后向其中添加锂化剂以使由通式(2)表示的膦硼烷锂化来进行。
[0052]
作为用于溶解由通式(2)表示的膦硼烷的溶剂,可以使用任何溶剂,而没有特别限制,只要它是对由通式(2)表示的膦硼烷和通过膦硼烷的锂化制造的锂化膦硼烷呈惰性的
溶剂即可。此类溶剂的实例包括四氢呋喃、n,n-二甲基甲酰胺、乙醚、叔丁基甲基醚、环戊基甲基醚、二恶烷、己烷和甲苯。这些溶剂可以单独使用或作为混合溶剂使用。
[0053]
在锂化膦硼烷的制备中,从反应性和制造率的角度来看,优选的是,在溶剂中由通式(2)表示的膦硼烷的浓度为1-80质量%,优选5-30质量%。
[0054]
作为在锂化膦硼烷的制备中使用的锂化剂,例如,使用有机锂化合物。有机锂化合物包括甲基锂、乙基锂、正丙基锂、仲丙基锂、正丁基锂、仲丁基锂和叔丁基锂。其中,从合适的碱性和充分的反应性的角度来看,正丁基锂是优选的。
[0055]
从经济效率和反应性的角度来看,优选的是,相对于由通式(2)表示的膦硼烷,要添加的锂化剂的量为1.0至1.5当量,优选1.0至1.2当量。
[0056]
从反应性和防止副反应的角度来看,优选的是,锂化的温度为-80℃至50℃,优选-20℃至20℃。
[0057]
锂化是通过将锂化剂添加到含有由通式(2)表示的膦硼烷的液体中来快速进行的,但为了完成锂化反应,根据需要,在锂化剂添加完成之后,可以相继地进行老化反应。
[0058]
锂化膦硼烷如上所述制备为溶液,并且可以按原样使用而无需分离,或根据需要,通过调节溶液浓度,用于第二步。
[0059]
由通式(3)表示的光学活性羟甲基膦硼烷可以通过熟知的方法制造。该方法的实例包括其中二烷基甲基膦硼烷被对映选择性地去质子化、然后被氧化的方法(参见日本专利特开第2010-209008号等)。
[0060]
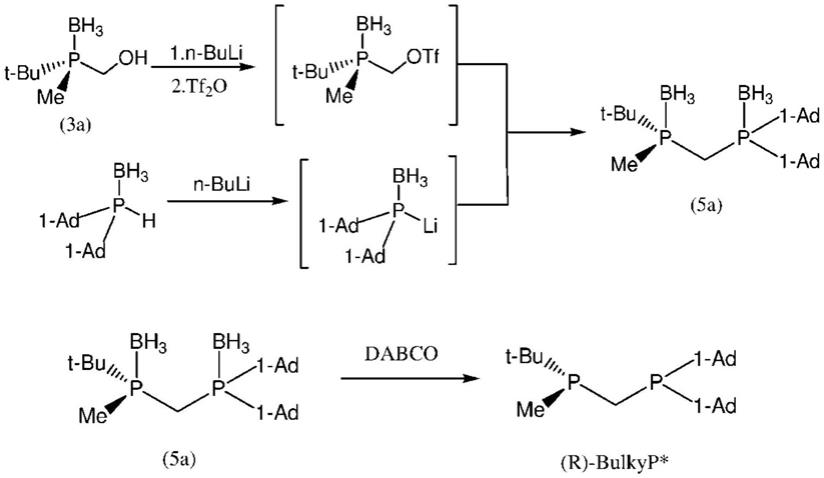
光学活性膦硼烷衍生物的制备可以通过使由通式(3)表示的光学活性羟甲基膦硼烷溶解在溶剂中并添加碱和羟基活化剂并且使其反应以将羟基转化为离去官能团来进行。
[0061]
碱的实例包括三乙胺、三丁胺、二异丙基乙胺、1,8-二氮杂双环[5.4.0]十一碳-7-烯、1,5-二氮杂双环[4.3.0]壬-5-烯、甲基锂、乙基锂、正丙基锂、仲丙基锂、正丁基锂、仲丁基锂和叔丁基锂。相对于由通式(3)表示的光学活性羟甲基膦硼烷,反应中使用的碱的量通常为1至3摩尔倍,并且优选1至2摩尔倍。
[0062]
羟基活化剂的实例包括甲磺酰氯、甲磺酸酐、对甲苯磺酰氯和对甲苯磺酸酐。相对于由通式(3)表示的光学活性羟甲基膦硼烷,反应中使用的羟基活化剂的量通常为1至5摩尔倍,并且优选1至2摩尔倍。
[0063]
反应中使用的溶剂没有特别限制,只要它对反应呈惰性即可,并且其实例包括四氢呋喃、n,n-二甲基甲酰胺、乙醚、叔丁基甲基醚、环戊基甲基醚、二恶烷、己烷和甲苯。这些溶剂可以单独使用或作为混合溶剂使用。
[0064]
反应的反应温度通常为-80℃至50℃,并且优选-80℃至30℃。反应时间通常为0.5小时或更久,并且优选1至8小时。
[0065]
由通式(4)表示的光学活性膦硼烷衍生物如上所述制备为溶液,并且可以按原样使用而无需分离,或根据需要,通过调节溶液浓度,用于第二步。
[0066]
第二步是使锂化膦硼烷与光学活性膦硼烷衍生物反应以获得由以下通式(5)表示的光学活性双膦基甲烷硼烷的步骤。
[0067]
[式5]
[0068][0069]
其中r1、r2和*与通式(1)中的相同。
[0070]
该反应可以通过将第一步中制备的锂化膦硼烷溶液与由通式(4)表示的光学活性膦硼烷衍生物溶液混合来进行。混合方法没有特别限制,但优选的是,因为反应容易控制,所以将锂化膦硼烷溶液滴入并与光学活性膦硼烷衍生物溶液混合。
[0071]
从反应性和经济效率的角度来看,优选的是,在锂化膦硼烷与由通式(4)表示的光学活性膦硼烷衍生物的摩尔比为0.5至3.0,尤其是1.0至1.5的条件下进行反应。
[0072]
从反应性和防止副反应的角度来看,优选的是,反应的反应温度为-80℃至50℃,尤其是-80℃至20℃。反应时间通常为0.5小时或更久并且优选1至8小时。
[0073]
反应结束后,根据需要,通过比如分液清洗、萃取、蒸馏、去溶剂化、柱色谱法或重结晶的常用方法进行纯化,可以获得由通式(5)表示的光学活性双膦基甲烷硼烷。
[0074]
第三步是用脱硼剂在溶剂中将第二步中获得的由通式(5)表示的光学活性双膦基甲烷硼烷脱硼以获得由通式(1)表示的目标光学活性双膦基甲烷的步骤。
[0075]
脱硼剂的实例包括n,n,n',n'-四甲基乙二胺(tmeda)、三乙二胺、(1,4-二氮杂双环[2.2.2]辛烷(dabco)、三乙胺、hbf4和三氟甲磺酸,但dabco是优选的。
[0076]
相对于由通式(5)表示的光学活性双膦基甲烷硼烷,脱硼反应中添加的脱硼剂的量通常为2至10当量并且优选3至5当量。
[0077]
作为反应中使用的溶剂,可以使用任何溶剂,而没有特别限制,只要它是能够溶解由通式(5)表示的光学活性双膦基甲烷硼烷且对双膦基甲烷硼烷和要制造的由通式(1)表示的光学活性双膦基甲烷呈惰性的溶剂即可。其实例包括乙酸乙酯、四羟基呋喃、n,n-二甲基甲酰胺、乙醚、叔丁基甲基醚、环戊基甲基醚、二恶烷、己烷和甲苯。这些溶剂可以单独使用或作为混合溶剂使用。
[0078]
从反应速度和获得的目标物质的纯度的角度来看,优选的是,脱硼反应的反应温度优选为-20℃至80℃,更优选20℃至80℃。脱硼反应的反应时间优选为10分钟或更久,并且更优选1至10小时。
[0079]
脱硼反应结束后,根据需要,通过比如分液清洗、萃取、结晶、蒸馏、升华或柱色谱法的常用方法进行纯化,可以获得由通式(1)表示的目标光学活性双膦基甲烷。
[0080]
由通式(1)表示的光学活性双膦基甲烷可以作为配体与过渡金属一起形成络合物。这种过渡金属络合物适用作不对称合成催化剂。不对称合成的实例包括脱氢氨基酸等的不对称加氢反应、伴随c-c键合或c-n键合的不对称偶联反应、不对称氢化硅烷化反应、不对称tsuji-trost反应和不对称硼化反应,比如对映选择性γ位硼取代反应。
[0081]
在有机化合物的合成中,该合成包括通过使用具有由通式(1)表示的光学活性双膦基甲烷作为配体的过渡金属络合物代替熟知的不对称催化剂,通过不对称催化剂进行不对称反应的步骤,在需要光学活性物质的领域中,可以高效地制造比如医药、农药、电子材料及其中间体的有机化合物。
[0082]
能够与由通式(1)表示的光学活性双膦基甲烷形成络合物的过渡金属的实例包括
铑、钌、铱、钯、镍、铁和铜。其中,铑和钯是优选的。
[0083]
用于与铑金属形成具有由通式(1)表示的光学活性双膦基甲烷作为配体的络合物的方法包括,例如,《实验化学指南(experiment chemistry guide book)》,第4版(由日本化学学会(the chemical society)编辑,丸善书店株式会社出版),第18卷,第327-353页)中描述的方法。具体地,可以通过使由通式(1)表示的光学活性双膦基甲烷与双(环辛烷-1,5-二烯)铑六氟锑酸盐、双(环辛烷-1,5-二烯)铑四氟硼酸盐等反应来制造铑络合物。
[0084]
用于与钯金属形成具有由通式(1)表示的光学活性双膦基甲烷作为配体的络合物的方法包括,例如,“y.uozumi和t.hayashi,《美国化学学会杂志(j.am.chem.soc.)》,1991,113,9887”中描述的方法。具体地,可以通过使由通式(1)表示的光学活性双膦基甲烷与π-烯丙基氯化钯反应来制造钯络合物。
[0085]
具有由通式(1)表示的光学活性双膦基甲烷作为配体的过渡金属络合物可以适当地用作不对称催化剂,特别是用于不对称加氢反应。这种情况下,过渡金属包括铑、钌和铱。其中,铑是优选的。
[0086]
可以应用不对称催化剂的反应包括使用熟知的不对称加氢催化剂的反应(例如,参见日本专利特开第2010-208993号、第2007-56007号、第2000-319288号、第2013-6787号和第2012-17288号)。
[0087]
实施例
[0088]
在下文中,将通过实施例的方式对本发明进行描述,但本发明并不受这些实施例的任何限制。
[0089]
(合成例1)
[0090]
《(r)-叔丁基(羟甲基)甲基膦硼烷(3a)》
[0091]
[式6]
[0092][0093]
将(s)-叔丁基甲基膦硼烷(5.90g,50mmol)和磁力搅拌棒放入安装有三通旋塞和隔垫的300ml四颈烧瓶中,并且重复抽真空和通入氩气多次以用氩气替换系统内部。添加脱水thf(100ml)以溶解(s)-叔丁基甲基膦硼烷;其后,将溶液冷却至-80℃,并在5分钟内滴加n-buli(1.57m己烷溶液,35.0ml,55mmol)。搅拌30分钟后,一次性加入多聚甲醛(4.50g,150mmol);并将所得物在剧烈搅拌下在2小时期间加热至室温。添加饱和氯化铵水溶液(50ml)以使反应停止;并且所得混合物用叔丁基甲基醚(50ml
×
两次)萃取。萃取液用饱和食盐水清洗,用无水硫酸钠干燥,过滤,并真空浓缩。通过硅胶柱色谱法(洗脱液:乙酸乙酯/己烷(1:3))纯化白色固体残余物,从而获得目标物质,为无色晶体(6.74g,产率:91%)。分析结果如下所示。
[0094]
mp 182℃至184℃(分解)
[0095]
[α]d
27-16.5(c=1.0,acoet)
[0096]
rf=0.37(acoet/己烷(1:3))
[0097]1h-nmr(500mhz,cdcl3)δ0.05-0.075(br s,3h),1.21(d,j
hp
=14.4hz,9h),1.27
(d,j
hp
=10.3hz,3h),2.02(br s,1h),3.95(d,j=13.2hz,1h),4.05(d,j=13.2hz,1h)
[0098]
13
c-nmr(125mhz,cdcl3)δ3.01(d,j
cp
=34.6hz),25.4,27.2(d,j
cp
=32.2hz),57.0(d,j
cp
=37.0hz)
[0099]
31
p-nmr(202mhz,cdcl3)δ28.2
[0100]
(实施例1)
[0101]
根据以下流程,合成(r)-二-1-金刚烷基膦基(叔丁基甲基膦基)甲烷((r)-bulkyp*)。
[0102]
[式7]
[0103][0104]
《第一步》
[0105]
将二-1-金刚烷基膦硼烷(1.581g,5mmol)放入安装有三通旋塞和隔垫的50ml双颈烧瓶中,并且重复抽真空和通入氩气以用氩气替换系统内部。添加脱水thf(25ml),并将所得混合物冷却至0℃;并在5分钟内滴加n-buli(1.42m己烷溶液,3.70ml,5.2mmol)。滴加后,将所得物在室温下搅拌30分钟,从而获得二-1-金刚烷基膦硼烷的锂化物质的溶液(液体a)。
[0106]
将(r)-叔丁基(甲基)羟甲基膦硼烷(740mg,5mmol)放入安装有三通旋塞和隔垫的100ml双颈烧瓶中,并且重复抽真空和通入氩气以用氩气替换系统内部。添加脱水乙醚(10ml),并将烧瓶浸入-80℃的低温浴中;并且在用磁力搅拌器搅拌下在5分钟期间滴加n-buli(1.42m己烷溶液,3.70ml,5.25mmol)。然后,用注射器在约10分钟期间添加三氟甲磺酸酐(0.86ml,5.25mmol);并将浴温升高到-30℃,并继续搅拌1小时,从而获得(r)-叔丁基(甲基)羟甲基膦硼烷的三氟甲磺酸酯溶液(液体b)。
[0107]
《第二步》
[0108]
通过套管连接两个含有液体a和液体b的烧瓶,并在约20分钟内将液体a逐滴转移到含有液体b的烧瓶中。在约2小时期间将浴温从-30℃升高到室温,并在该温度下继续搅拌过夜。
[0109]
反应混合物中的溶剂通过蒸发器除去;将水(20ml)添加到所得残余物中并充分搅拌,并且其后通过玻璃过滤器(4g)进行抽滤。将所得固体物质用水(5ml
×
两次)和甲醇(3ml
×
两次)清洗并真空干燥,从而获得白色粉末(1.75g)。将该粗产物通过柱色谱法(wako凝胶c300,110g,二氯甲烷/己烷(3:1))纯化,从而获得(r)-硼酸根(叔丁基甲基膦基)硼酸根(二-1-金刚烷基)膦基甲烷(5a)(1.20g,产率:54%)。分析结果如下所示。
[0110]
mp大约280℃
[0111]
[α]d
24
=-8.0(c 1.02,cdcl3)
[0112]
rf=0.56(acoet/己烷(1:5))
[0113]1h-nmr(500mhz,cdcl3)δ0.2-1.0(br m,6h),1.23(d,3j
hp
=13.8hz,9h),1.57(d,2j
hp
=10.3hz,3h),1.70-1.80(m,12h),1.82-1.90(m,2h),1.97-2.18(m,18h)
[0114]
13
c-nmr(125mhz,cdcl3)δ6.1(dd,j
cp
=20.9,14.9hz),6.6(d,j
cp
=32.2hz),25.3,28.1-28.2(m),30.1(d,j
cp
=35.8hz),36.4,36.5,37.6,37.8,37.9,38.9(d,j
cp
=22.7hz)
[0115]
31
p-nmr(200mhz,cdcl3)δ32.6,40.9
[0116]
《第三步》
[0117]
将(r)-硼酸根(叔丁基甲基膦基)硼酸根(二-1-金刚烷基)膦基甲烷(223mg,0.5mmol)和dabco(337mg,3mmol)放入安装有三通旋塞和隔垫的10ml双颈烧瓶中,并重复抽真空和通入氩气以用氩气替换系统内部。添加脱气甲苯(2.5ml);其后,将烧瓶浸入80℃的油浴中;并在用磁力搅拌器搅拌下使所得物反应3小时。其后,将烧瓶直接连接到蒸发器并除去溶剂。添加4ml甲醇并充分搅拌约10分钟;并且其后,在3g玻璃过滤器上过滤固体物质,并用甲醇(3ml
×
两次)清洗。所得物进一步用乙酸乙酯(2ml
×
两次)清洗,并且其后真空干燥,从而获得呈白色粉末状的(r)-二-1-金刚烷基膦基(叔丁基甲基膦基)甲烷((r)-bulkyp*)(195mg,产率:93%)。分析结果如下所示。
[0118]
mp大约265℃
[0119]
rf=0.85(acoet/己烷(1:5))
[0120]1h-nmr(500mhz,cdcl3)δ1.02(d,2j
hp
=3.5hz,3h),1.06(d,3j
hp
=11.5hz,9h),1,55-1.62(m,2h),1.68-1.73(m,12h),1.82-1.87(m,6h),1.92-1.99(m,12h)
[0121]
13
c-nmr(125mhz,cdcl3)δ7.1(dd,j=19.5,6.6hz),11.4(dd,j=31.0,22.7hz),26.6(d,j=13.1hz),28.1(m),28.7(m),36.2(m),36.7(m),37.1,40.9(d,j=10.7hz),41.3(dd,j=9.6,3.6hz)
[0122]
31
p-nmr(202mhz,cdcl3)δ-13.2(d,j
pp
=114hz),13.5(d,j
pp
=114hz)
[0123]
将在上面获得的(r)-bulkyp*白色粉末在25℃的空气中静置24小时,并且其后再次进行1h-nmr、
13
c-nmr和
31
p-nmr的测量以进行确认,但未观察到杂质,表明(r)-bulkyp*在空气中稳定。
[0124]
(实施例2)
[0125]
根据以下流程,合成(r)-bulkyp*的铑络合物。
[0126]
[式8]
[0127][0128]
将[rh(cod)2]sbf6(111mg,0.20mmol)放入安装有三通旋塞和隔垫的20ml双颈烧瓶
中,并重复抽真空和通入氩气以用氩气替换系统内部,并且其后通过添加脱气二氯甲烷(6ml)溶解所得物。单独地将(r)-bulkyp*(92mg,0.22mmol)放入安装有三通旋塞和隔垫的10ml双颈烧瓶中,并重复抽真空和通入氩气以用氩气替换系统内部,并且其后添加脱气thf(2ml)以溶解(r)-bulkyp*。将该溶液用注射器萃取,在充分搅拌下在约10分钟期间滴加在上面制备的[rh(cod)2]sbf6的二氯甲烷溶液中。1小时后,用蒸发器除去溶剂;并将1.5ml乙酸乙酯添加到所得残余物中并通过搅拌将内容物充分混合。过滤沉积的橙色沉淀物,并用乙酸乙酯(0.5ml
×
三次)清洗并真空干燥(151mg,87%)。
[0129]
将所获得的产物在氩气气氛下溶解于二氯甲烷(0.50ml)中;并用注射器将2.0ml乙酸乙酯一次性添加至所得溶液中。将获得的均相溶液在冰箱中冷却,并过滤沉积的晶体,并用二氯甲烷/乙酸乙酯(1:4)的混合溶剂清洗,并且其后真空干燥,从而获得(r)-bulkyp*的铑络合物的橙色晶体(123mg,产率:71%)。分析结果如下所示。
[0130]
mp 230℃(分解)
[0131]1h-nmr(500mhz,cdcl3)δ1.18(d,3j
hp
=15.5hz,9h),1.71(d,2j
hp
=8.6hz,3h),1.73-2.20(m,30h),2.20-2.33(m,4h),2.38-2.54(m,4h),3.15-3.30(m,2h),5.07(br s,1h),5.11(br s,1h),5.69(br s,1h),5.79(br s,1h)
[0132]
13
c-nmr(125mhz,cdcl3)δ9.3(d,j=20.3hz),25.9(t,j=18.5hz),26.6(d,j=3.6hz),28.3(d,j=8.4hz),28.4(d,j=8.4hz),28.9,29.1,30.9,31.6,33.2(dd,j=17.3,4.1hz),36.3,36.4,40.4,40.8,41.3(d,j=4.8hz),43.4,91.0(m),91.7(m),97.4(m),100.7(m)
[0133]
31
p-nmr(202mhz,cdcl3)δ-14.6(dd,j
pp
=124hz,j
prh
=53hz),-30.5(br d,j
pp
=124hz)
[0134]
(实施例3-1至3-12)
[0135]
《不对称加氢反应》
[0136]
将在实施例2中制备的(r)-bulkyp*的铑络合物(0.005mmol,4.3mg)和0.5mmol的由下式(a1)表示的底物加入100ml耐压反应管中。反应管为不锈钢管,并连接到氢气罐。用氢气置换反应管五次后,充入1个大气压的氢气(日本精工株式会社(japan fine products co.,ltd.)制造,99.99999%)。通过使用注射器,向耐压反应管中添加3ml脱气甲醇。然后,将反应管中的氢气压力设定为3个大气压(在实施例3至5中,为1个大气压)。在加氢反应在搅拌下进行表1所指示的反应时间后,将反应管中剩余的氢气放出,并用蒸发器将反应液浓缩,以获得残余物。通过快速色谱法(sio2,乙酸乙酯/己烷(3:1))纯化残余物以获得由式(a2)表示的产物。产物的绝对构型和ee值通过比较保留时间与先前报告的值来确定。结果示于表1中。
[0137]
然后,通过1h、
13
c和
31
p的每种nmr测量确认,在反应期间,形成了其中(r)-bulkyp*与一价铑离子以1:1配位的金属络合物。
[0138]
[式9]
[0139][0140]
[表1]
[0141][0142]
1)压力为1个大气压
[0143]
在式中,me是甲基;ph是苯基;ac是乙酰基。
[0144]
(实施例4-1至4-3)
[0145]
《不对称加氢反应》
[0146]
将实施例2中制备的(r)-bulkyp*的铑络合物(0.005mmol,4.3mg)和0.5mmol的由下式(a3)表示的底物加入100ml的耐压反应管中,然后进行与实施例3-1至3-12中相同的程序以获得由式(a4)表示的产物。产物的绝对构型和ee值通过比较保留时间与先前报告的值来确定。结果示于表2中。
[0147]
然后,通过1h、
13
c和
31
p的每种nmr测量确认,在反应期间,形成了其中(r)-bulkyp*与一价铑离子以1:1配位的金属络合物。
[0148]
[式10]
[0149][0150]
[表2]
[0151][0152]
如表1和表2中所指示,显然具有由通式(1)表示的光学活性双膦基甲烷作为配体的过渡金属络合物在不对称加氢反应中表现出高对映选择性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1