一种雷诺嗪中间体杂质的制备方法与流程
1.本发明属于化合物制备技术领域,尤其涉及一种雷诺嗪中间体杂质的制备方法。
背景技术:
2.雷诺嗪是一种部分脂肪酸氧化酶抑制剂,通过阻滞心肌晚钠离子电流,降低细胞内钙水平超载,从而使舒张期左心室壁压降低,减少心肌氧耗。雷诺嗪通过抑制钠和钾离子通道电流起作用。这种作用是通过抑制钠离子通道的峰值和延迟而获得的,其依次增加了心肌功能。据报道雷诺嗪在钠通道中的作用是组织特异性以及频率和电压依赖性的,已证明雷诺嗪在心动过速方面更有效。同样,雷诺嗪以11.5μmol/l的抑制浓度抑制延迟的整流器钾电流,从而延长了心室动作电位的持续时间。同样,已经显示出雷诺嗪对l型钙通道具有小的活性,使其成为弱的直接血管舒张剂,并且对房室结传导的影响最小。因此,雷诺嗪的作用是通过抑制延迟的整流器钾电流和抑制内向钠电流之间的结合而获得的。该药用于治疗慢性心绞痛。
3.雷诺嗪由吉利德(gilead sciences)公司研发,持证商为gilead sciences inc,首先于2006年1月27日获美国食品药品管理局(fda)批准上市,之后于2008年7月9日获欧洲药物管理局(ema)批准上市,由吉利德公司在美国和欧洲销售,商品名为ranexa。ranexa为口服缓释片,每片含有500mg或1000mg雷诺嗪。临床推荐剂量为每日500mg两次或每日1000mg两次。
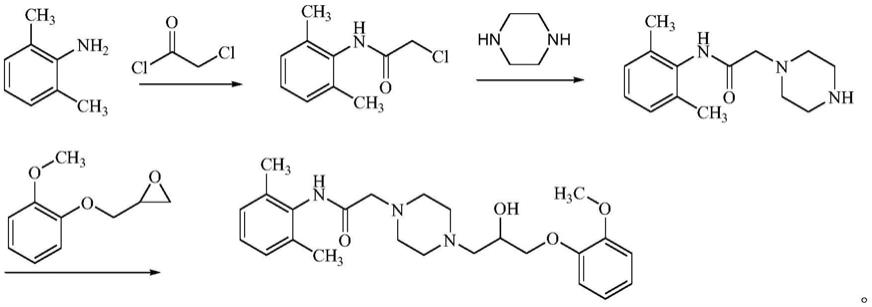
4.最早报道雷诺嗪的合成路线的专利有us4567264,ep0483932a1,wo2006008753a1等。根据原研化合物专利us4567264报道的路线,该产品以2,6-二甲基苯胺作为起始原料,和氯乙酰氯酰胺化反应生成n-)2,6-二甲基苯基)氯乙酰胺;之后和哌嗪发生亲核取代反应生成n-(2,6-二甲基苯基)-2-(哌嗪-1-基)乙酰胺,然后再与愈创木酚缩水甘油醚反应得到雷诺嗪成品。其具体反应路线如下:
[0005][0006]
药物开发过程中杂质的规范研究是重要的一个环节,将产品中的杂质控制在一个安全合理的限度范围之内,是药物质量研究中的一项重要工作,同时质量标准的建立需要一定量的标准品。
技术实现要素:
[0007]
有鉴于此,本发明的目的在于提供一种雷诺嗪中间体杂质的制备方法,该方法简单,产品收率高。
[0008]
本发明提供了一种雷诺嗪中间体杂质的制备方法,包括以下步骤:
[0009]
在碱性物质和催化剂作用下,将n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺在溶剂中进行反应,得到雷诺嗪中间体杂质;
[0010]
所述雷诺嗪中间体杂质具有式ⅰ结构:
[0011][0012]
优选地,所述n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺的摩尔比为1:1~1:2。
[0013]
优选地,所述溶剂选自n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二甲基亚砜和1,4-二氧六环中的一种或多种。
[0014]
优选地,所述碱性物质选自碳酸铯、碳酸钾、碳酸钠、碳酸氢钠、氢氧化钾和氢氧化钠中的一种或多种。
[0015]
优选地,n-(2,6-二甲基苯基)氯乙酰胺与所述碱性物质的摩尔比为1:1~1:4;
[0016]
n-(2,6-二甲基苯基)氯乙酰胺与所述催化剂的摩尔比为1:0.01~1:1。
[0017]
优选地,所述催化剂选自碘化钾、碘化钠、四丁基溴化铵和四丁基醋酸铵中的一种或多种。
[0018]
优选地,所述n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺反应的温度为60~100℃。
[0019]
优选地,所述n-(2,6-二甲基苯基)氯乙酰胺由2,6-二甲基苯胺和氯乙酰氯发生酰胺化反应制得。
[0020]
优选地,将反应产物降温,滴加水的过程中,反应产物先溶清,然后析出固体,滴加完毕后再室温搅拌55~65min,过滤,滤饼洗涤至中性,干燥,得到雷诺嗪中间体杂质。
[0021]
本发明提供了一种雷诺嗪中间体杂质,具有式ⅰ结构:
[0022][0023]
本发明提供的方法采用n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺为原料,在碱性和催化剂作用下,即可获得具有式ⅰ结构的雷诺嗪中间体杂质。该方法简单,目标产物收率高,且纯度高,可用于雷诺嗪工艺研发、生产、质量标准建立、质量控制环节,为雷诺嗪的用药安全性提供技术支持。实验结果表明:本发明提供的方法所制得的雷诺嗪中间体杂质纯度最高达99.23%,收率最高达54.36%,产品纯度较高,产量充足,可满足分析日常所需的方法建立和杂质控制。
附图说明
[0024]
图1为本发明雷诺嗪中间体杂质的化学反应方程式图;
[0025]
图2为本发明实施例1中雷诺嗪中间体杂质的核磁共振氢谱图;
[0026]
图3为本发明实施例1中雷诺嗪中间体杂质的质谱图;
[0027]
图4为本发明实施例1中雷诺嗪的高效液相色谱图。
具体实施方式
[0028]
本发明提供了一种雷诺嗪中间体杂质的制备方法,包括以下步骤:
[0029]
在碱性物质和催化剂作用下,将n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺在溶剂中进行反应,得到雷诺嗪中间体杂质;
[0030]
所述雷诺嗪中间体杂质具有式ⅰ结构:
[0031][0032]
为了开展雷诺嗪的工艺研究和质量研究,提高雷诺嗪的用药安全性,研究人员首先要获得足够量的杂质对照品,在研究过程中发现,通过文献的合成方法,此二聚体杂质的存在量甚微,难以通过分离反应液获得足够量的杂质对照品,而改变文献报道的工艺条件亦无法获得足够多的杂质对照品的量。目前市面上报道关于雷诺嗪合成方法的文献较多,但关于此杂质的合成方法没有报道。
[0033]
本发明提供的方法采用n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺为原料,在碱性和催化剂作用下,即可获得具有式ⅰ结构的雷诺嗪中间体杂质。该方法简单,目标产物收率高,且纯度高,可用于雷诺嗪工艺研发、生产、质量标准建立、质量控制环节,为雷诺嗪的用药安全性提供技术支持。
[0034]
在本发明中,所述n-(2,6-二甲基苯基)氯乙酰胺优选由2,6-二甲基苯胺和氯乙酰氯发生酰胺化反应制得。
[0035]
所述2,6-二甲基苯胺和氯乙酰氯发生酰胺化反应在碱性物质存在下反应;酰胺化反应采用的碱性物质优选选自三乙胺、碳酸钠、碳酸氢钠、碳酸钾和n,n-二异丙基乙胺中的一种或多种,更优选选自三乙胺。
[0036]
所述酰胺化反应在溶剂中进行;酰胺化反应采用的溶剂优选选自四氢呋喃、1,4-二氧六环、乙二醇二甲醚和二氯甲烷中的一种或多种。
[0037]
所述2,6-二甲基苯胺和氯乙酰氯的摩尔比优选为1:1~1:2,更优选为1:1.5;所述2,6-二甲基苯胺和氯乙酰氯酰胺化反应的温度优选为60~100℃。本发明采用tlc板检测反应进度,待反应原料消耗完,反应即可结束。
[0038]
酰胺化反应结束后,水洗2~3次,合并水相,萃取,得到的有机相洗涤至中性,干燥,过滤,得到的母液浓缩干后加入甲基叔丁基醚,室温打浆,抽滤,滤饼干燥至恒重,得到n-(2,6-二甲基苯基)氯乙酰胺。
[0039]
在本发明中,所述n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺的摩尔比优选为1:1~2,更优选为1:1.5。
[0040]
n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺反应在溶剂中进行;所述溶剂优选选自n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二甲基亚砜和1,4-二氧六环中的一种或多种。
[0041]
n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺反应在碱性物质和催化剂作用下进行;所述碱性物质优选选自碳酸铯、碳酸钾、碳酸钠、碳酸氢钠、氢氧化钾和氢氧化钠中的一种或多种,更优选为碳酸钾和/或碳酸钠。所述催化剂优选选自碘化钾、碘化钠、四丁基溴化铵和四丁基醋酸铵中的一种或多种,更优选为碘化钾。
[0042]
在本发明中,n-(2,6-二甲基苯基)氯乙酰胺与所述碱性物质的摩尔比为1:1~1:4;
[0043]
n-(2,6-二甲基苯基)氯乙酰胺与所述催化剂的摩尔比为1:0.01~1:1。
[0044]
在本发明中,所述n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺反应的温度为60~100℃,优选为75~80℃;具体实施例中,所述n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺反应的温度为80℃,时间为4h。
[0045]
本发明优选将n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺的反应产物降温,滴加水的过程中,反应产物先溶清,然后析出固体,滴加完毕后再室温搅拌55~65min,过滤,滤饼洗涤至中性,干燥,得到雷诺嗪中间体杂质。
[0046]
具体实施例中,室温搅拌的时间为60min;干燥的温度为60℃。
[0047]
本发明在雷诺嗪合成路线的基础上制备了此杂质,为雷诺嗪原料药质量的有效控制夯实了基础。对于雷诺嗪中的杂质:n-(2,6-二甲基苯基)-2-(2,6-二甲基苯基氨基)乙酰胺的制备具有重要的意义,它可以用于雷诺嗪合成中杂质的定性及定量分析等质量研究,从而有助于提高雷诺嗪的产品质量,并为降低雷诺嗪的用药风险提供重要的指导意义。
[0048]
为了进一步说明本发明,下面结合实施例对本发明提供的一种雷诺嗪中间体杂质及其制备方法进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0049]
实施例1
[0050]
图1为本发明雷诺嗪中间体杂质的化学反应方程式图;
[0051]
步骤1:
[0052]
在250ml三口瓶中加入60ml二氯甲烷,12.10ggg2,6-二甲基苯胺和15.15g三乙胺,降温至0℃,然后滴加14.56g的氯乙酰氯,滴加过程中控制温度≤10℃,滴完保持温度≤10℃搅拌1个小时。反应完加入纯化水洗100ml
×
2次,水相合并用50ml二氯甲烷萃取一次,合并有机相用饱和碳酸氢钠溶液洗至ph=7,无水硫酸钠干燥,过滤,母液浓缩干后加入100ml甲基叔丁基醚,室温打浆1h,抽滤,滤饼60℃干燥至恒重后得到针状白色晶体17.89g,收率90.81%。
[0053]
步骤2:
[0054]
在250ml三口瓶中加入10.00gn-(2,6-二甲基苯基)氯乙酰胺,9.08g2,6-二甲基苯胺,50ml n,n-二甲基甲酰胺,13.8g碳酸钾和0.83g碘化钾,加热至80℃反应,采用tlc板检测反应进度。待反应原料消耗完毕后结束反应,将得到的反应产物降至室温,滴加200ml纯化水,反应液先溶清,而后析出固体,加毕室温搅拌1h,过滤,滤饼用纯化水洗涤中性后60℃干燥至恒重后得到8.48g淡黄色固体,收率60.18%。
[0055]
图4为本发明实施例1中雷诺嗪的高效液相色谱图;对其分析见表1:
[0056]
表1实施例1中雷诺嗪的高效液相色谱图的数据分析
[0057][0058]
实施例2:
[0059]
步骤1:
[0060]
在250ml三口瓶中加入60ml四氢呋喃,12.10ggg2,6-二甲基苯胺和15.15g三乙胺,降温至0℃,然后滴加14.56g的氯乙酰氯,滴加过程中控制温度≤10℃,滴完保持温度≤10℃搅拌1个小时。反应完加入100ml二氯甲烷和纯化水分液,有机相用100ml纯化水,合并水相用50ml二氯甲烷萃取一次,水相丢弃,有机相合并用饱和碳酸氢钠溶液洗至ph=7,无水硫酸钠干燥,过滤,母液浓缩干后加入100ml甲基叔丁基醚,室温打浆1h,抽滤,滤饼60℃干燥至恒重后得到针状白色晶体18.10g,收率91.87%。
[0061]
步骤2:
[0062]
在250ml三口瓶中加入10.00gggn-(2,6-二甲基苯基)氯乙酰胺,9.08ggg2,6-二甲基苯胺,50ml 1,4-二氧六环,13.8g碳酸钾和0.83g碘化钾,加热至80℃反应,采用tlc板检测反应进度。待反应原料消耗完毕后结束反应,将得到的反应产物降至室温,滴加200ml纯化水,反应液先溶清,而后析出固体,加毕室温搅拌1h,过滤,滤饼用纯化水洗涤中性后60℃干燥至恒重后得到8.18g淡黄色固体,收率58.01%。
[0063]
实施例3:
[0064]
与实施例1的区别在于,步骤1中将碱换为n,n-二异丙基乙胺,步骤2中将碱换为碳酸钠;
[0065]
实施例4:
[0066]
与实施例1的区别在于,步骤1中将反应溶剂换为氯仿,步骤2中将反应溶剂换为n-甲基吡咯烷酮。
[0067]
方法和结果:
[0068]
色谱条件:
[0069]
色谱柱:ymc-pack ods-a c18;流动相:0.05mol/l磷酸二氢钾缓冲溶液(含0.2%的三乙胺):乙腈=48:52(v/v);流速:1.0ml/min;检测波长215nm;进样量:10μl。在此色谱条件下,色谱柱的理论塔板数按雷诺嗪计算应不低于3 000。
[0070]
表2实施例1~4的检测结果:
[0071]
样品批次纯度收率
实施例199.23%54.36%实施例298.89%53.24%实施例398.60%52.16%实施例498.38%52.64%
[0072]
以上结果显示,本发明所制得的雷诺嗪中间体杂质纯度最高达99.23%,收率最高达54.36%,产品纯度较高,产量充足,可满足分析日常所需的方法建立和杂质控制。
[0073]
由以上实施例可知,本发明提供的方法采用n-(2,6-二甲基苯基)氯乙酰胺和2,6-二甲基苯胺为原料,在碱性和催化剂作用下,即可获得具有式ⅰ结构的雷诺嗪中间体杂质。该方法简单,目标产物收率高,且纯度高,可用于雷诺嗪工艺研发、生产、质量标准建立、质量控制环节,为雷诺嗪的用药安全性提供技术支持。实验结果表明:本发明提供的方法所制得的雷诺嗪中间体杂质纯度最高达99.23%,收率最高达54.36%,产品纯度较高,产量充足,可满足分析日常所需的方法建立和杂质控制。
[0074]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1