靶向CDK4或CDK6的共价抑制剂及其应用
靶向cdk4或cdk6的共价抑制剂及其应用
技术领域
1.本发明涉及靶向cdk4或cdk6的共价抑制剂及其应用,属于共价药物设计技术领域。
背景技术:
2.细胞周期蛋白依赖性激酶(cyclin dependent kinases,cdks)是一类丝氨酸/苏氨酸蛋白激酶,属于gmgc家族,在调节细胞周期进程中发挥着重要作用。cdks必须通过与各自对应的细胞周期蛋白(cyclins)结合形成蛋白激酶复合物才具有活性,二者皆是细胞进行分裂必不可少的组分。cdks目前已发现20种亚型,依据功能的不同分为两大类:负责调控细胞周期的主要包括cdk1、cdk2、cdk4、cdk6等;与转录调节相关的主要包括cdk7、cdk8、cdk9、cdk10、cdk11及cdk12等。还有一些cdks在其他功能上发挥着不可或缺的作用,如cdk5是调节神经元活性,参与代谢的重要激酶,cdk16与精子形成密切相关。
3.其中,cdk4/6即细胞周期依赖性激酶4(cdk4)或细胞周期依赖性激酶6(cdk6),通过与细胞周期蛋白d结合,调节细胞周期从g1期转换到s期。cdk4与cdk6蛋白结构与功能相似,有71%的同源氨基酸,均能与cyclin d结合形成异源二聚体,磷酸化人视网膜母细胞瘤蛋白(rb),然后rb释放磷酸化前与其紧密结合的转录因子e2f,e2f活化后促进细胞周期相关基因的转录,使得细胞由g1期进入s期,细胞周期增殖得以进行。另外,cdk4/6也能通过磷酸化其他转录因子抑制衰老,促进增殖。在体内,cdk4/6活性受内源性cdks抑制剂ink4家族所调控。ink4家族由p15/ink4b,p16/ink4a,p18/ink4c和p19/ink4d四个成员组成,它们均能与细胞周期蛋白d结合,竞争性抑制cdk4/6-cyclin d二聚体的形成。据统计,约90%的人类癌症出现cyclin d-cdk4/6-ink4-rb信号通路的异常活化。该信号通路的失常,将导致肿瘤细胞无限增殖,因此,以该通路组成作为抗肿瘤治疗的分子靶标,特别是通过抑制癌细胞中过度表达的cdk4/6使得其阻滞在g1期并最终导致肿瘤细胞凋亡的策略被认为是目前阻断cdk4/6信号通路的最优选择。
4.目前,已有不少选择性cdk4/6抑制剂的抗肿瘤作用被临床验证。这其中包括已上市的cdk4/6抑制剂帕博西尼(palbociclib),瑞博西尼(ribociclib),abemaciclib。随着细胞周期调控研究的不断深入,越来越多的选择性cdk4/6抑制剂进入临床研究或临床前研究,如trilaciclib(g1t-28),flx-925,lerociclib(g1t-38),以及我国药企恒瑞公司自主研发的cdk6抑制剂shr-6390等,其分子结构式如下:
[0005][0006]
其中,palbociclib(帕博西尼)是来自辉瑞公司的全球首款靶向cdk4/6的口服小分子抑制剂,于2015年2月获得美国fda快速审批通道上市,其抗肿瘤活性较好,对cdk4及cdk6激酶的ic50为9-15nm,被批准用于治疗er+/her2-的晚期或转移性乳腺癌患者。研究发现,palbociclib能抑制多种乳腺癌细胞系,特别是er+的乳腺癌细胞系。在临床ii期研究(paloma-1)中,palbociclib联合芳香酶抑制剂来曲唑用于治疗165例绝经不适合手术的er+/her2-乳腺癌患者,较来曲唑单药相比,无进展生存期(pfs)由10.2月提高到20.2月,且无明显副作用。基于此,nccn指南推荐帕博西尼联合芳香酶抑制剂作为治疗er+/her2-晚期乳腺癌的一线治疗方案。
[0007]
靶向共价抑制剂(targeted covalent inhibitor,tci)是一种设计成共价结合特定分子靶标从而抑制其生物学功能的化合物,结合过程常为不可逆的。与传统药物相比,共价抑制剂具有显著的优势,如强靶亲和力、延长作用时间、减少服药剂量及频次等。另外,共价抑制剂由于暴露时间长,可以实现完全抑制,因而与非共价抑制剂相比,更不易产生耐药性。但是,设计一种共价药物可能是具有挑战性的,因为现有的知识无法提供最佳的方法,一些目前可用的共价药物的共价性质是在其开发之后发现的。随着研究人员对共价药物越来越深入的了解,利用现有的非共价抑制剂进行结构改造,在合适部位连接上亲电弹头,组成新共价抑制剂的设计策略被发展起来。当前,共价抑制剂的设计多以活性位点上的催化或非催化半胱氨酸为靶点,α,β-不饱和羰基等迈克尔受体作为亲电试剂进攻半胱氨酸残基中的巯基,而以其他类型的激酶氨基酸为作用靶点研究较少。而目前未见将palbociclib改造为共价药物的报道。
技术实现要素:
[0008]
针对以上缺陷,本发明解决的技术问题是提供一种靶向cdk4或cdk6的共价抑制剂,以palbociclib为基础,进行改造优化,得到一种新型共价抑制剂。
[0009]
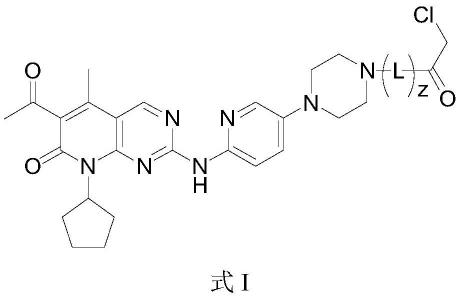
本发明靶向cdk4或cdk6的共价抑制剂,结构式为式i所示:
[0010][0011]
其中,z为0或1;
[0012]
l为m1为1或2,n1为1~5中的任一整数,m2为1或2,n2为1~5中的任一整数;*为帕博西尼哌嗪环中的n。
[0013]
作为其中一种实施方式,z为0。
[0014]
作为另一种实施方式,z为1。
[0015]
在具体的实施例中,n1为1,2,3或5;n2为1或2。
[0016]
本发明还提供本发明所述的化合物在制备靶向cdk4或cdk6的共价抑制剂中的应用。
[0017]
本发明还提供本发明所述的化合物在制备治疗肿瘤药物中的应用。
[0018]
在本发明的一些实施例中,所述肿瘤为肺癌或乳腺癌。
[0019]
本发明还提供一种用于治疗肿瘤的药物组合物。
[0020]
本发明用于治疗肿瘤的药物组合物,含有治疗有效量的本发明所述化合物和药学上可接受的载体。
[0021]
本发明首次提出在palbociclib化学结构中处于溶剂区且对活性影响不大哌嗪环上安装一个特定的共价弹头α-氯代酰胺,与palbociclib结合口袋的外周有一保守的氨基酸残基thr107共价偶联,改造后的化合物并未改变原分子的母核结构,结合方式不会受到太大影响;且能够通过共价作用增加小分子与靶蛋白间的结合能力,提高抑制活性,同时降低对正常细胞的毒性。
附图说明
[0022]
图1为化合物c-13与cdk6靶蛋白的结合模式。
[0023]
图2为化合物c-13与palbociclib对lo2细胞的抑制率。
[0024]
图3为化合物c-13作用于mda-mb-231细胞周期分布图。
[0025]
图4为palbociclib作用于mda-mb-231细胞周期分布图。
[0026]
图5为化合物c-13作用于mda-mb-231细胞的annexin v/pi双染实验结果图。
[0027]
图6为化合物c-13作用于mda-mb-231细胞24h后cdk4/6及prb蛋白含量变化图。
[0028]
图7为化合物c-13不同给药浓度下克隆的形成图。
[0029]
图8为肿瘤体积测量示意图。
[0030]
图9为给药时间与肿瘤体积的变化图。
[0031]
图10为给药时间与小鼠体重的变化图。
具体实施方式
[0032]
目前多数共价抑制剂的研究主要靶向于半胱氨酸巯基,本发明通过对palbociclib与cdk6共晶结构(2euf)进行分析,发现在palbociclib结合口袋的外周有一保守的氨基酸残基thr107,据此,提出了一种新的共价抑制剂,以palbociclib作为导引头,在其哌嗪环仲氮上安装一个亲电弹头,共价修饰thr107。
[0033]
本发明的化合物,结构式为式i所示:
[0034][0035]
其中,z为0或1;
[0036]
l为m1为1或2,n1为1~5中的任一整数,m2为1或2,n2为1~5中的任一整数;*为帕博西尼哌嗪环中的n。
[0037]
作为其中一种实施方式,z为0。当z为0时,表示没有连接基团l,帕博西尼哌嗪环中的n直接与α-氯代酰胺中的羰基碳连接,其结构式为:
[0038][0039]
作为另一种实施方式,z为1。
[0040]
在具体的实施例中,n1为1,2,3或5;n2为1或2。
[0041]
具体的,以下为本发明优选的化合物结构式:
[0042][0043]
本发明还提供本发明所述的化合物在制备靶向cdk4或cdk6的共价抑制剂中的应用。
[0044]
本发明化合物,可作为靶向cdk4或cdk6的共价抑制剂,通过α-氯代酰胺亲电弹头与苏氨酸共价偶联,这种作用模式的优势是:首先,化合物分子与靶蛋白通过共价偶联,大大提高了结合能力,会带来更强的生物活性,其次,并未改变抑制剂分子的母核结构,结合模式不会受到影响,仍然保持较好的选择性;再次,目前多数共价抑制剂的研究主要靶向于半胱氨酸巯基,而本发明靶向于苏氨酸,为除半胱氨酸以外的其它氨基酸残基的共价抑制剂的研究提供了新思路。
[0045]
本发明还提供本发明所述的化合物在制备治疗肿瘤药物中的应用。
[0046]
本发明化合物,可以用于制备治疗肿瘤的药物中。在本发明的一些实施例中,所述肿瘤为肺癌或乳腺癌。
[0047]
本发明还提供一种用于治疗肿瘤的药物组合物。
[0048]
本发明用于治疗肿瘤的药物组合物,含有治疗有效量的本发明所述化合物和药学上可接受的载体。本发明所述药学上可接受的载体为药学领域常规的药物载体,比如,稀释剂、赋形剂、填充剂等。
[0049]
下面结合实施例对本发明的具体实施方式做进一步的描述,并不因此将本发明限制在所述的实施例范围之中。
[0050]
实施例1
[0051]
以帕博西尼(palbociclib)为底物的重要中间体的合成:
[0052]
分别通过在碱性条件下将palbociclib与溴乙酸叔丁酯或丙烯酸叔丁酯偶联,然后酸解生成化合物pd-3或pd-5。
[0053][0054]
条件和试剂:a.1.5eq溴乙酸叔丁酯,10.0eq dipea,dcm,r.t,4h;b.3.0eq丙烯酸叔丁酯,3.0eq dbu,dcm,r.t,5h;c.dcm/tfa=3:1,r.t,5h。
[0055]
以α-氯代酰胺为弹头c类共价化合物的合成路线:
[0056][0057][0058]
条件和试剂:g.2.0eq氯乙酰氯,3.0eq三乙胺,thf,0℃-r.t,5h。
[0059]
具体中间体结构及反应步骤如下所示:
[0060]
中间体pd-4的合成:
[0061][0062]
将pabociclib(111.88mg,0.25mmol)溶于30ml dcm中,加入dipea(0.412ml,0.296mmol),溴乙酸叔丁酯(0.054ml,0.375mmol)室温下搅拌5h。tlc监测反应直至反应完全。后处理:先减压浓缩除去溶剂dcm,再加入40ml水,用ea萃取(3
×
40ml)水层,合并有机相,用饱和食盐水洗三次然后用无水硫酸钠干燥,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体115mg,产率82%。1h nmr(400mhz,cdcl3)δ8.89(s,1h),8.15(d,j=9.2hz,1h),8.11(d,j=2.8hz,1h),7.33(dd,j=9.1,3.0hz,1h),3.30
–
3.23(m,4h),3.20(s,2h),2.84
–
2.74(m,4h),2.55(s,3h),2.42
–
2.31(m,5h),2.09
–
2.02(m,2h),1.92
–
1.86(m,2h),1.48(s,9h),1.26(s,3h).
[0063]
中间体pd-5的合成:
[0064][0065]
将pd-4中间体溶于6ml dcm中,往其中加入2ml三氟乙酸,室温下搅拌5h。停止反应后减压浓缩,然后加入dcm(6
×
20ml)减压蒸馏得到绿色固体pd-5,产率约为90%。
[0066]
中间体pd-2的合成:
[0067][0068]
将pabociclib(44.75mg,0.1mmol)溶于15ml dcm中,加入dbu(0.044ml,0.296mmol),丙烯酸叔丁酯(0.045ml,0.312mmol)室温下搅拌5h。tlc监测反应直至反应完全。后处理:先减压浓缩除去溶剂dcm,再加入30ml水,用etoac萃取(3
×
25ml)水层,合并有机相,用饱和食盐水洗三次然后用无水硫酸钠干燥,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体54mg,产率94%。1h nmr(400mhz,cdcl3)δ8.87(s,1h),8.16(d,j=9.1hz,1h),8.08(d,j=2.7hz,1h),7.32(dd,j=13.1,6.6hz,1h),3.27
–
3.17(m,4h),2.74(t,j=7.3hz,2h),2.70
–
2.61(m,4h),2.55(s,3h),2.46(t,j=7.3hz,2h),2.42
–
2.31(m,5h),2.10
–
2.02(m,2h),1.92
–
1.87(m,2h),1.45(s,9h),1.26(s,3h).
[0069]
中间体pd-3的合成:
[0070][0071]
将pd-2中间体溶于6ml dcm中,往其中加入2ml三氟乙酸,室温下搅拌5h。停止反应后减压浓缩,然后加入dcm(6
×
20ml)减压蒸馏得到绿色固体pd-3,产率约为90%。
[0072]
中间体8a的合成:
[0073][0074]
将pd-5(50mg,1.0eq)溶于15ml dmf中,溶液澄清,往其中加入n-叔丁氧羰基-1,2-乙二胺(0.119mmol,1.2eq),hatu(45mg,1.2eq),dipea(0.048ml,3.0eq),室温下反应12h。tlc监测反应直至反应完全。后处理:停止反应后加入30ml水,用etoac萃取(3
×
25ml)水层,合并有机相,用饱和食盐水洗三次后用无水硫酸钠干燥,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体9mg,产率14%。1h nmr(400mhz,cdcl3)δ8.85(s,1h),8.18(d,j=9.1hz,1h),8.05(d,j=2.8hz,1h),7.33(dd,j=9.1,2.9hz,1h),4.04(s,2h),3.50(s,4h),3.28
–
3.19(m,4h),3.12(s,2h),2.77
–
2.68(m,4h),2.55(s,3h),2.41
–
2.30(m,5h),2.10
–
2.03(m,2h),1.92
–
1.86(m,2h).
[0075]
中间体8b的合成:
[0076][0077]
中间体8b的合成方法与8a相同,可参照其操作方法,仅将原料更换为n-叔丁氧羰基-1,3-丙二胺(0.119mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体25mg,产率38%。1h nmr(400mhz,cdcl3)δ8.87(s,1h),8.17(d,j=9.1hz,1h),8.07(d,j=2.8hz,1h),7.33(dd,j=9.1,2.9hz,1h),3.35(t,j=6.5hz,2h),3.30
–
3.23(m,4h),3.18(t,2h),3.11(s,2h),2.79
–
2.70(m,4h),2.55(s,3h),2.41
–
2.30(m,5h),2.11
–
2.02(m,2h),1.89
–
1.87(m,2h),1.69
–
1.66(m,2h),1.44(s,9h),1.26(s,3h).
[0078]
中间体8c的合成:
[0079][0080]
中间体8c的合成方法与8a相同,可参照其操作方法,仅将原料更换为n-叔丁氧羰基-1,4-丁二胺(0.119mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体18mg,产率27%。1h nmr(400mhz,cdcl3)δ8.92(s,1h),8.17(d,j=9.1hz,1h),8.08(d,j=2.8hz,1h),7.32(dd,j=9.1,2.9hz,1h),3.33(t,j=6.1hz,2h),3.25
–
3.21(m,4h),3.16
–
3.09(m,4h),2.76
–
2.70(m,4h),2.55(s,3h),2.40
–
2.33(m,5h),2.10
–
2.05(m,2h),1.93
–
1.85(m,2h),1.59
–
1.52(m,4h),1.43(s,9h),1.26(s,3h).
[0081]
中间体8d的合成:
[0082][0083]
中间体8d的合成方法与8a相同,可参照其操作方法,仅将原料更换为n-叔丁氧羰基-1,6-己二胺(0.119mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体35mg,产率50%。1h nmr(400mhz,cdcl3)δ8.90(s,1h),8.17(d,j=9.1hz,1h),8.08(d,j=2.8hz,1h),7.34(dd,j=9.1,2.9hz,1h),3.34
–
3.14(m,8h),3.10(s,2h),2.79
–
2.70(m,4h),2.55(s,3h),2.41
–
2.31(m,5h),2.10
–
2.04(m,2h),1.93
–
1.85(m,2h),1.73
–
1.66(m,2h),1.56
–
1.51(m,2h),1.43(s,9h),1.37
–
1.34(m,4h),1.26(s,3h).
[0084]
中间体9a的合成:
[0085][0086]
中间体9a的合成方法与8a相同,可参照其操作方法,仅将原料更换为[2-(2-氨基乙氧基)乙基]氨基甲酸叔丁酯(0.119mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体25mg,产率37%。
[0087]
中间体9b的合成:
[0088][0089]
中间体9b的合成方法与8a相同,可参照其操作方法,仅将原料更换为2-(2-(2-氨基乙氧基)乙氧基)乙基氨基甲酸叔丁酯(0.119mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体20mg,产率29%。1h nmr(400mhz,cdcl3)δ8.87(s,1h),8.17(d,j=9.7hz,1h),8.07(d,j=2.8hz,1h),7.34(dd,j=9.1,2.9hz,1h),3.75
–
3.56(m,8h),3.53(t,j=5.2hz,4h),3.26
–
3.20(m,4h),3.12(s,2h),2.55(s,3h),2.41
–
2.33(m,5h),2.10
–
2.04(m,2h),1.93
–
1.86(m,2h),1.44(s,9h),1.26(s,3h).
[0090]
中间体10a的合成:
[0091][0092]
将pd-3(50mg,1.0eq)溶于15ml dmf中,溶液澄清,往其中加入n-叔丁氧羰基-1,2-乙二胺(0.116mmol,1.2eq),hatu(44.1mg,1.2eq),dipea(0.048ml,3.0eq),室温下反应12h。tlc监测反应直至反应完全。后处理:停止反应后加入30ml水,用etoac萃取(3
×
25ml)水层,合并有机相,用饱和食盐水洗三次后用无水硫酸钠干燥,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体23mg,产率36%。1h nmr(400mhz,cdcl3)δ8.82(s,1h),8.18(d,j=9.1hz,1h),8.05(d,j=2.6hz,1h),7.33(dd,j=9.1,2.8hz,1h),3.41
–
3.34(m,2h),3.30
–
3.21(m,6h),2.78
–
2.62(m,6h),2.55(s,3h),2.44(t,j=6.1hz,2h),2.40
–
2.30(m,5h),2.11
–
2.03(m,2h),1.93
–
1.85(m,2h),1.43(s,9h),1.26(s,3h).
[0093]
中间体10b的合成:
[0094][0095]
中间体10b的合成方法与10a相同,可参照其操作方法,仅将原料更换为n-叔丁氧羰基-1,3-丙二胺(0.116mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体18mg,产率28%。1h nmr(400mhz,cdcl3)δ8.87(s,
1h),8.17(d,j=9.1hz,1h),8.08(d,j=2.8hz,1h),7.33(dd,j=9.1,2.9hz,1h),3.30(t,j=12.4,6.1hz,2h),3.26
–
3.21(m,4h),3.17(t,j=12.3,6.1hz,2h),2.78
–
2.66(m,6h),2.55(s,3h),2.45(t,j=6.3hz,2h),2.39
–
2.30(m,5h),2.11
–
2.03(m,2h),1.91
–
1.87(m,2h),1.72
–
1.66(m,2h),1.42(s,9h),1.26(s,3h).
[0096]
中间体10c的合成:
[0097][0098]
中间体10c的合成方法与10a相同,可参照其操作方法,仅将原料更换为n-叔丁氧羰基-1,4-丁二胺(0.116mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体36mg,产率54%。1h nmr(400mhz,cdcl3)δ8.87(s,1h),8.17(d,j=8.6hz,1h),8.08(d,j=2.8hz,1h),7.34(dd,j=9.1,3.0hz,1h),3.29
–
3.19(m,6h),3.15
–
3.10(m,2h),2.77
–
2.67(m,6h),2.55(s,3h),2.44(t,j=6.1hz,2h),2.40
–
2.30(m,5h),2.13
–
1.98(m,4h),1.93
–
1.85(m,2h),1.72
–
1.67(m,2h),1.42(s,9h),1.26(s,3h).
[0099]
中间体10d的合成:
[0100][0101]
中间体10d的合成方法与10a相同,可参照其操作方法,仅将原料更换为n-叔丁氧羰基-1,6-己二胺(0.116mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体37mg,产率54%。
[0102]
中间体11a的合成:
[0103][0104]
中间体11a的合成方法与10a相同,可参照其操作方法,仅将原料更换为[2-(2-氨基乙氧基)乙基]氨基甲酸叔丁酯(0.116mmol,1.2eq),后处理方法也相同,减压蒸馏后,用
dcm/meoh体系作为流动相进行柱层析分离得到绿色固体28mg,产率42%。
[0105]
中间体11b的合成:
[0106][0107]
中间体11b的合成方法与10a相同,可参照其操作方法,仅将原料更换为2-(2-(2-氨基乙氧基)乙氧基)乙基氨基甲酸叔丁酯(0.116mmol,1.2eq),后处理方法也相同,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到绿色固体43mg,产率64%。1h nmr(400mhz,cdcl3)δ8.87(s,1h),8.19(d,j=9.1hz,1h),8.08(d,j=2.8hz,1h),7.34(dd,j=9.1,2.9hz,1h),3.62
–
3.55(m,6h),3.53
–
3.43(m,4h),3.31
–
3.20(m,6h),2.79
–
2.67(m,6h),2.55(s,3h),2.45(t,j=6.3hz,2h),2.42
–
2.33(m,5h),2.10
–
2.03(m,2h),1.92
–
1.84(m,2h),1.44(s,9h),1.26(s,3h).
[0108]
具体目标化合物结构及反应步骤如下所示:
[0109]
目标化合物c-1的合成:
[0110][0111]
将中间体8a(20mg,1.0eq)溶于3ml dcm中,往其中加入1ml三氟乙酸,室温下搅拌3h。停止反应后减压浓缩,然后加入dcm(3
×
10ml)减压蒸馏得到棕色油状物。将该油状物中溶解于15ml thf中,加入tea(0.013ml,3.0eq)置于冰浴条件下搅拌5min,同时氮气保护。然后将氯乙酰氯(0.006ml,2.0eq)在此条件下缓慢滴加,溶液由黄绿色变成橙红色。30min后将该反应移置于室温下继续反应5小时。tlc监测反应直至反应完全。后处理:先减压浓缩除去溶剂thf,再加入30ml水,用ea萃取(3
×
30ml)水层,合并有机相,用饱和食盐水洗三次后用无水硫酸钠干燥,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体9mg,产率46%。1h nmr(400mhz,cdcl3)δ8.85(s,1h),8.18(d,j=9.1hz,1h),8.05(d,j=2.8hz,1h),7.33(dd,j=9.1,2.9hz,1h),4.04(s,2h),3.56
–
3.44(m,4h),3.28
–
3.19(m,4h),3.12(s,2h),2.77
–
2.68(m,4h),2.55(s,3h),2.41
–
2.30(m,5h),2.10
–
2.03(m,2h),1.92
–
1.86(m,2h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.63,171.21,166.81,161.42,158.07,157.23,155.55,145.18,143.31,141.76,136.60,130.80,126.10,113.62,107.80,61.40,54.07,53.39,49.57,42.54,40.60,38.79,31.53,28.09,25.76,13.96.hrms(esi
+
):calculated for c
30h38
cln9o4[m+h]
+
624.2735,found 624.2788。
[0112]
目标化合物c-2的合成:
[0113][0114]
目标化合物c-2的合成方法与c-1相同,可参照其操作方法,中间体10a(23mg,1.0eq),氯乙酰氯(0.007ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体8mg,产率23%。1h nmr(400mhz,cdcl3)δ8.73(s,1h),8.11(d,1h),7.96(d,j=2.8hz,1h),7.26(dd,j=2.9hz,1h),3.98
–
3.95(m,2h),3.40
–
3.35(m,4h),3.19
–
3.15(m,4h),2.72
–
2.65(m,6h),2.48
–
2.47(s,3h),2.43(t,j=6.1hz,2h),2.31
–
2.28(m,5h),2.03
–
1.98(m,2h),1.83
–
1.77(m,2h),1.26(s,3h).hrms(esi
+
):calculated for c
31h40
cln9o4[m+h]
+
638.2892,found 638.2961。
[0115]
目标化合物c-3的合成:
[0116][0117]
目标化合物c-3的合成方法与c-1相同,可参照其操作方法,中间体8b(25mg,1.0eq),氯乙酰氯(0.007ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体15mg,产率61%。1h nmr(400mhz,cdcl3)δ8.80(s,1h),8.15(d,j=13.6hz,1h),8.02(d,1h),7.34(d,j=7.8hz,1h),4.06(s,2h),3.41
–
3.32(m,4h),3.29
–
3.20(m,4h),3.13(s,2h),2.80
–
2.71(m,4h),2.58
–
2.52(s,3h),2.41
–
2.31(m,5h),2.10
–
2.03(m,2h),1.91
–
1.85(m,2h),1.74
–
1.66(m,4h),1.26(s,3h).hrms(esi
+
):calculated for c
31h40
cln9o4[m+h]
+
638.2892,found 638.2957。
[0118]
目标化合物c-4的合成:
[0119][0120]
目标化合物c-4的合成方法与c-1相同,可参照其操作方法,中间体10b(28mg,1.0eq),氯乙酰氯(0.008ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体10mg,产率36%。1h nmr(400mhz,cdcl3)δ8.84(s,1h),8.18(d,j=9.1hz,1h),8.06(d,j=2.8hz,1h),7.33(dd,1h),4.04(s,2h),3.38
–
3.30
(m,4h),3.24
–
3.20(m,4h),2.76
–
2.70(m,6h),2.56
–
2.54(s,3h),2.47(t,j=6.1hz,2h),2.39
–
2.33(m,5h),2.09
–
2.03(m,2h),1.90
–
1.86(m,2h),1.70
–
1.67(m,2h),1.26(s,3h).
13
c nmr(101mhz,dmso)δ202.92,171.58,166.31,161.23,159.03,158.72,155.23,144.72,143.91,142.56,135.81,129.68,125.14,115.59,107.03,54.52,53.38,52.71,48.77,43.13,37.22,36.54,33.72,31.77,29.48,28.03,25.57,14.08.hrms(esi
+
):calculated for c
32h42
cln9o4[m+h]
+
652.3048,found 652.3112。
[0121]
目标化合物c-5的合成:
[0122][0123]
目标化合物c-5的合成方法与c-1相同,可参照其操作方法,中间体8c(40mg,1.0eq),氯乙酰氯(0.01ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体9mg,产率22.8%。1h nmr(400mhz,cdcl3)δ8.86(s,1h),8.18(d,j=9.1hz,1h),8.06(d,j=2.8hz,1h),7.34(dd,j=9.1,2.9hz,1h),4.04(s,2h),3.39
–
3.32(m,4h),3.26
–
3.19(m,4h),3.10(s,2h),2.77
–
2.70(m,4h),2.57
–
2.52(s,3h),2.41
–
2.32(m,4h),2.10
–
2.03(m,1h),1.92
–
1.85(m,1h),1.64
–
1.56(m,4h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.64,169.96,166.00,161.42,158.08,157.24,155.55,145.18,143.32,141.78,136.55,130.78,126.05,113.60,107.77,61.59,54.08,53.38,49.58,42.68,39.49,38.47,31.53,28.08,27.28,26.77,25.76,13.96.hrms(esi
+
):calculated for c
32h42
cln9o4[m+h]
+
652.3048,found 652.3115。
[0124]
目标化合物c-6的合成:
[0125][0126]
目标化合物c-6的合成方法与c-1相同,可参照其操作方法,中间体10c(24mg,1.0eq),氯乙酰氯(0.007ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体10mg,产率42%。1h nmr(400mhz,cdcl3)δ8.81(s,1h),8.19(d,j=9.1hz,1h),8.04(d,j=2.8hz,1h),7.35(dd,j=9.1,2.9hz,1h),4.00(s,2h),3.37
–
3.26(m,4h),3.25
–
3.18(m,4h),2.77
–
2.67(m,6h),2.55(s,3h),2.45(t,j=6.1hz,2h),2.39
–
2.31(m,5h),2.11
–
2.04(m,2h),1.91
–
1.86(m,2h),1.77
–
1.73(m,4h),1.26(s,3h).hrms(esi
+
):calculated for c
33h44
cln9o4[m+h]
+
666.3205,found 666.3288。
[0127]
目标化合物c-7的合成:
[0128][0129]
目标化合物c-7的合成方法与c-1相同,可参照其操作方法,中间体8d(40mg,1.0eq),氯乙酰氯(0.011ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体9mg,产率23%。1h nmr(400mhz,cdcl3)δ8.84(s,1h),8.17(d,j=9.1hz,1h),8.05(d,j=2.7hz,1h),7.34(dd,j=9.1,2.9hz,1h),4.04(s,2h),3.34
–
3.27(m,4h),3.26
–
3.20(m,4h),3.11(s,2h),2.80
–
2.70(m,4h),2.55(s,3h),2.40
–
2.30(m,5h),2.10
–
2.05(m,2h),2.03
–
1.96(m,4h),1.72
–
1.66(m,2h),1.57
–
1.54(m,2h),1.38
–
1.35(m,2h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.63,169.54,165.87,161.40,158.06,157.23,155.54,145.17,143.29,141.76,136.51,130.77,126.10,113.63,107.77,61.55,54.05,53.32,49.52,42.70,39.62,38.76,31.52,29.63,29.22,28.08,26.38,26.28,25.75,13.95.hrms(esi
+
):calculated for c
34h46
cln9o4[m+na]
+
702.3361,found 702.3257。
[0130]
目标化合物c-8的合成:
[0131][0132]
目标化合物c-8的合成方法与c-1相同,可参照其操作方法,中间体10d(24mg,1.0eq),氯乙酰氯(0.006ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体4mg,产率17%。1h nmr(400mhz,cdcl3)δ8.82(s,1h),8.19(d,j=9.1hz,1h),8.04(d,j=2.6hz,1h),7.35(dd,j=9.0,2.8hz,1h),4.03(s,2h),3.33
–
3.15(m,8h),2.81
–
2.63(m,6h),2.55(s,3h),2.44(t,2h),2.40
–
2.30(m,5h),2.10
–
2.03(m,2h),1.92
–
1.86(m,2h),1.56
–
1.46(m,4h),1.40
–
1.32(m,4h),1.26(s,3h).hrms(esi
+
):calculated for c
35h48
cln9o4[m+h]
+
694.3518,found 694.3588。
[0133]
目标化合物c-9的合成:
[0134]
[0135]
目标化合物c-9的合成方法与c-1相同,可参照其操作方法,中间体9a(25mg,1.0eq),氯乙酰氯(0.007ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体15mg,产率61%。1h nmr(400mhz,cdcl3)δ8.85(s,1h),8.18(d,j=9.1hz,1h),8.05(d,j=2.8hz,1h),7.33(dd,j=9.1,3.0hz,1h),4.05(s,2h),3.62
–
3.57(m,4h),3.54
–
3.48(m,4h),3.26
–
3.20(m,4h),3.13(s,2h),2.77
–
2.71(m,4h),2.55(s,3h),2.40
–
2.32(m,5h),2.10
–
2.04(m,2h),1.91
–
1.86(m,2h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.65,170.14,166.00,161.42,158.07,157.23,155.55,145.18,143.29,141.78,136.51,130.78,126.04,113.62,107.78,70.01,69.16,61.60,54.06,53.33,49.57,42.67,39.65,38.68,31.53,28.09,25.76,13.96.hrms(esi
+
):calculated for c
32h42
cln9o5[m+h]
+
668.2997,found 668.3061。
[0136]
目标化合物c-10的合成:
[0137][0138]
目标化合物c-10的合成方法与c-1相同,可参照其操作方法,中间体11a(28mg,1.0eq),氯乙酰氯(0.008ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体18mg,产率65%。1h nmr(400mhz,cdcl3)δ8.85(s,1h),8.19(d,j=9.1hz,1h),8.06(d,j=2.8hz,1h),7.34(dd,j=9.1,2.9hz,1h),4.03(s,2h),3.59
–
3.53(m,4h),3.49
–
3.43(m,4h),3.25
–
3.18(m,4h),2.77
–
2.67(m,6h),2.55(s,3h),2.46(t,j=6.3hz,2h),2.40
–
2.33(m,5h),2.09
–
2.03(m,2h),1.91
–
1.86(m,2h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.67,172.36,166.06,161.42,158.08,157.24,155.55,145.17,143.30,141.78,136.41,130.77,125.95,113.63,107.77,70.03,69.10,54.06,53.88,52.41,49.51,42.66,39.58,38.82,32.55,31.53,28.09,25.77,13.96.hrms(esi
+
):calculated for c
33h44
cln9o5[m+na]
+
704.3154,found 704.3129。
[0139]
目标化合物c-11的合成:
[0140][0141]
目标化合物c-11的合成方法与c-1相同,可参照其操作方法,中间体9b(25mg,1.0eq),氯乙酰氯(0.007ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体12mg,产率49%。1h nmr(400mhz,cdcl3)δ8.82(s,1h),8.17(d,1h),8.03(d,j=2.7hz,1h),7.34(dd,j=9.1,2.9hz,1h),4.05(s,2h),3.65
–
3.55(m,8h),3.54
–
3.46(m,4h),3.25
–
3.19(m,4h),3.11(s,2h),2.77
–
2.71(m,4h),2.55(s,3h),2.39
–
2.32(m,5h),2.09
–
2.04(m,2h),1.91
–
1.86(m,2h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.65,169.91,166.04,161.41,158.05,157.21,155.54,145.14,143.32,141.75,136.32,130.82,126.17,113.69,100.00,70.41,70.20,70.00,69.45,61.53,53.24,49.53,42.66,39.52,38.74,31.52,29.69,28.09,25.77,13.96.hrms(esi
+
):calculated for c
34h46
cln9o6[m+na]
+
734.3260,found 734.3309。
[0142]
目标化合物c-12的合成:
[0143][0144]
目标化合物c-12的合成方法与c-1相同,可参照其操作方法,中间体11b(42mg,1.0eq),氯乙酰氯(0.011ml,2.0eq),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体13mg,产率31%。1h nmr(400mhz,cdcl3)δ8.84(s,1h),8.18(d,j=9.1hz,1h),8.06(d,j=2.8hz,1h),7.34(dd,j=9.1,2.9hz,1h),4.06(d,j=10.1hz,2h),3.63
–
3.55(m,6h),3.54
–
3.50(m,2h),3.49
–
3.40(m,4h),3.28
–
3.17(m,4h),2.78
–
2.65(m,6h),2.55(s,3h),2.44(t,j=12.9,6.6hz,2h),2.40
–
2.29(m,5h),2.11
–
2.04(m,2h),1.91
–
1.87(m,2h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.65,172.22,166.06,161.42,158.08,157.24,155.54,145.14,143.37,141.78,136.35,130.78,126.01,113.65,107.79,70.34,70.13,69.39,54.04,53.88,52.38,49.47,42.68,39.51,38.90,32.57,31.53,28.09,25.77,13.96.hrms(esi
+
):calculated for c
35h48
cln9o6[m+h]
+
726.3416,found 726.3475。
[0145]
目标化合物c-13的合成:
[0146][0147]
将原料palbociclib(30mg,0.067mmol)溶于15ml dcm中,加入tea(0.03ml,0.2mmol)置于冰浴条件下搅拌5min,同时氮气保护。然后将氯乙酰氯(0.015ml,0.134mmol)在此条件下缓慢滴加,溶液由黄绿色变成橙红色。30min后将该反应移置于室温下继续反应5小时。tlc监测反应直至反应完全。后处理:先减压浓缩除去溶剂dcm,再加入30ml水,用ea萃取(3
×
30ml)水层,合并有机相,用饱和食盐水洗三次后用无水硫酸钠干燥,减压蒸馏后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体27mg,产率75%。1h nmr(400mhz,cdcl3)δ8.92(s,1h),8.22(d,j=9.0hz,1h),8.11(d,j=2.6hz,1h),7.35(dd,j=
9.1,2.8hz,1h),4.14(s,2h),3.85
–
3.72(m,4h),3.27
–
3.17(m,4h),2.55(s,3h),2.42
–
2.33(m,5h),2.12
–
2.03(m,2h),1.93
–
1.85(m,2h),1.26(s,3h).
13
c nmr(101mhz,cdcl3)δ202.61,165.20,161.39,158.04,157.21,155.55,145.89,143.04,141.72,137.42,130.92,126.88,113.59,107.90,54.10,50.04,49.68,46.16,41.98,40.73,31.52,28.10,25.77,13.97.hrms(esi
+
):calculated for c
26h30
cln7o3[m+h]
+
524.2099,found 524.2158。
[0148]
实施例2化合物对肿瘤细胞的抗增殖活性
[0149]
1、试剂与材料
[0150]
含有10%胎牛血清的dmem完全培养基,mtt,0.25%胰酶,10%pbs缓冲液,96孔板。用于测定细胞存活率的化合物均由实施例1中方法合成,dmso作溶剂配成20mg/ml的浓度贮存于4℃冰箱。
[0151]
本实验所用的细胞株包括:mda-mb-231(人乳腺癌细胞),mda-mb-453(人乳腺癌细胞),mcf-7(人乳腺癌细胞)和h1299(人肺癌细胞)。
[0152]
2、方法步骤
[0153]
取处于对数生长期的肿瘤细胞用0.25%胰酶消化,离心,接着用含10%胎牛血清的dmem完全培养基稀释,以3-5
×
103/孔的细胞密度接种于96孔板,每孔培养基100μl。接种完成后,将96孔板置于co2孵箱,37℃、5%co2条件下培养24小时。待肿瘤细胞贴壁生长到一定密度后给药,将用dmso配制的目标化合物溶液用培养基倍比稀释,设置给药浓度为20、10、5、2.5、1.25、0.625μm,每个浓度均为3个复孔。给药后继续培养72小时,吸除每孔中的液体,然后每孔加入mtt 20μl(5mg/ml)放置于培养箱4小时,吸出每孔中的液体,加入150μl dmso,充分摇晃10~15min使甲瓒充分溶解。然后使用酶标仪选择490nm和570nm作为测定波长,测定各孔的吸光度(od值)。根据公式:细胞抑制率%=(对照组od值-用药组od值)/对照细胞od值
×
100%,从而计算出各个目标化合物与其对应的肿瘤细胞的体外增殖抑制率,再通过ic
50
计算软件求得半数抑制浓度(ic
50
,μm)。为减少实验误差,重复三次实验。
[0154]
3、结果分析
[0155]
实施例1中化合物对4中人源性肿瘤细胞株的体外增殖抑制作用结果如表1。
[0156]
表1目标化合物的体外增殖的抑制活性ic
50
(μm)
[0157][0158]
由表1可知,以α-氯代酰胺为弹头的c系列化合物有多个分子具有体外抗增殖活性。其中化合物c-13对mda-mb-231、mda-mb-453及h1299细胞株的活性远远大于阳性药palbociclib。
[0159]
实施例3化合物对cdk4与cdk6的激酶活性研究
[0160]
为了检测该化合物与cdk4及cdk6蛋白的结合能力,我们从其中选择了活性较好的9个化合物进行纯度测定后,对cdk6/cycd3及cdk4/cycd3进行激酶活性的测定。
[0161]
化合物浓度的配制:
[0162]
9个受试化合物在cdk6及cdk4激酶上进行筛选,测试浓度为200nm或者20nm,2个浓度,复孔检测;化合物c-13从900nm起始,3倍稀释,10个浓度,单孔检测;同时以palbociclib作为阳性对照化合物。将事先配制的浓度为10mm的12个受试化合物在384孔板中稀释成100倍终浓度的100%dmso溶液,然后使用分液器echo 550向最终的384孔板转移250nl 100倍终浓度的化合物。
[0163]
激酶反应过程:
[0164]
1、用1
×
kinase buffer配制2.5倍终浓度的激酶溶液。
[0165]
2、在受试化合物孔和阳性对照孔分别加10μl的2.5倍终浓度的激酶溶液;在阴性对照孔中加10μl的1
×
kinase buffer。
[0166]
3、1000rpm离心30秒,反应板振荡混均匀后室温孵育10min。
[0167]
4、用1
×
kinase buffer配制5/3倍终浓度的atp和kinase substrate 8的混合溶液。
[0168]
5、加入15μl的5/3倍终浓度的atp和底物的混合溶液,开始反应。
[0169]
6、将384孔板1000rpm离心30秒,振荡混匀后室温孵育150min。
[0170]
7、加入30μl终止检测液停止激酶反应,1000rpm离心30秒,振荡混匀。
[0171]
8、用caliper ez reader读取转化率。
[0172]
9、数据分析计算。
[0173]
具体测试测试结果如下表2:
[0174]
表2受试化合物分子在200nm和20nm对cdk4及cdk6的抑制率(%)
[0175][0176]
根据激酶活性测试结果,我们发现在浓度为20nm时,9个受试化合物对cdk4及cdk6的抑制率基本均超过50%,这充分表明我们设计合成的以α-氯代酰胺为弹头的共价化合物具有靶向cdk6与cdk4的良好的活性。在20nm时,化合物c-4、c-5对cdk6激酶的抑制率高于化合物c-13,化合物c-3、c-5、c-6、c-10对cdk4激酶的抑制率高于c-13,此外,化合物c-5对cdk6及cdk4蛋白的抑制率均略大于化合物c-13,但是综合对人肿瘤细胞的体外增殖抑制作用结果来看,化合物c-5对多种肿瘤细胞的ic
50
值均远小于化合物c-13,这可能是因为化合物c-5的溶解性能或膜渗透性较差。
[0177]
由于化合物c-13的肿瘤细胞抑制活性最佳且对cdk6及cdk4激酶具有较好的抑制率,我们将其从900nm起始,3倍稀释,10个浓度,单孔检测,以palbociclib作为阳性对照,将浓度的log值作为x轴,百分比抑制率为y轴,通过分析软件graphpad prism 5拟合量效曲线,测得化合物c-13的ic
50
值,其结果见表3和表4。
[0178]
表3化合物c-13对cdk4及cdk6抑制率与浓度的关系
[0179][0180]
表4化合物c-13对cdk4及cdk6激酶的ic
50
值
[0181] 化合物c-13(nm)cdk6(cyclin d3)14cdk4(cyclin d3)6.1
[0182]
从表中我们可以发现化合物c-13对cdk6(cyclin d3)蛋白的ic
50
值为14nm,对cdk4(cyclin d3)蛋白的ic
50
值为6.1nm,虽弱于文献报道的cdk6(10.27nm)、cdk4(3.44nm),但对多种肿瘤细胞株的抗肿瘤细胞增殖作用大于阳性对照药物palbociclib,从另一方面反映了化合物c-13对cdk6及cdk4蛋白的较强结合能力,也进一步表明了该类分子的有效性及设计思路的可行性。
[0183]
实施例4化合物c-13结合模式的预测及初步验证
[0184]
为了预测化合物c-13与cdk6蛋白的结合模式,保持palbociclib在cdk6蛋白中结合模式不变,通过add fragment模块构建化合物c-13的初始构象,然后利用amber12对该体系进行能量最小化,最终得到目标构象。化合物c-13与cdk6靶蛋白的结合模式见图1。
[0185]
将含palbociclib片段的小分子化合物c-13对接到靶蛋白中,从预测的结合模式可知,保留了氨基嘧啶和氨基吡啶骨架,以及形成的四个关键氢键,并未改变palbociclib母核与cdk6蛋白的结合方式,从而有利于维持良好的酶活性和细胞活性。新活性分子c-13能较好地与cdk6靶蛋白活性位点结合,通过不断变换linker的长度能够使头部的α-氯代酰胺弹头靠近苏氨酸thr107。合适的链长对于化合物活性至关重要:linker太长柔性较大,易摆动产生复杂的热力学运动,与苏氨酸结合的几率随之减小;linker太短,头部的亲电基团不能靠近氨基酸残基与之作用。此外,亲电弹头与目标氨基酸残基的相互作用受多种因素的影响,包括弹头的亲电能力,靶蛋白活性口袋氨基酸残基的亲核性能等,最终能否触发反应形成共价作用与亲电弹头的选择密不可分。
[0186]
为了初步验证化合物c-13化合物是否通过共价键产生抑制活性发挥关键作用,我们将化合物c-13中的α-氯代酰胺基团替换为乙酰胺基团或丙酰胺基团,其结构式如下:
[0187][0188]
化合物d-1的合成:
[0189]
化合物d-1的合成方法与c-13相同,可参照其操作方法,palbociclib(30mg,0.067mmol),乙酰氯(0.010ml,0.134mmol),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体28mg,产率85%。1h nmr(400mhz,cdcl3)δ8.87(s,1h),8.21(d,j=9.1hz,1h),8.08(d,j=2.6hz,1h),7.35(dd,j=9.1,3.0hz,1h),3.86
–
3.76(m,2h),3.70
–
3.63(m,2h),3.23
–
3.13(m,4h),2.55(s,3h),2.42
–
2.31(m,5h),2.16(s,3h),2.11
–
2.03(m,2h),1.92
–
1.85(m,2h).13c nmr(101mhz,cdcl3)δ202.62,169.04,161.40,158.11,157.29,155.57,145.80,143.18,141.80,137.29,130.79,126.67,113.59,107.71,54.15,50.06,49.75,46.12,41.24,31.53,28.07,25.74,21.32,13.96.hrms(esi
+
):calculated for c
26h31
n7o3[m+h]
+
490.2488,found 490.2557。
[0190]
化合物d-2的合成:
[0191]
化合物d-2的合成方法与c-13相同,可参照其操作方法,palbociclib(30mg,0.067mmol),丙酰氯(0.012ml,0.134mmol),后处理方法也相同,减压浓缩后,用dcm/meoh体系作为流动相进行柱层析分离得到黄绿色固体31mg,产率92%。1h nmr(400mhz,cdcl3)δ8.92(s,1h),8.20(d,j=9.1hz,1h),8.11(d,j=2.8hz,1h),7.34(dd,j=9.1,3.0hz,1h),3.82(t,2h),3.67(t,2h),3.20
–
3.13(m,4h),2.55(s,3h),2.43
–
2.37(m,7h),2.10
–
2.04(m,2h),1.92
–
1.86(m,2h),1.19(t,3h).13c nmr(101mhz,cdcl3)δ202.63,172.37,161.40,158.12,157.31,155.57,145.76,143.22,141.82,137.25,130.74,126.58,113.57,107.66,54.16,50.08,49.78,45.22,41.36,31.53,28.06,26.47,25.74,13.95,9.46.hrms(esi
+
):calculated for c
27h33
n7o3[m+h]
+
504.2645,found 504.2710。
[0192]
为了验证化合物c-13弹头还原后的化合物d-1及d-2是否能产生体外抗增殖活性,我们通过mtt实验测定了其对以下四种人肿瘤细胞株的作用。此外,为了进一步测试还原后的化合物对cdk6和cdk4蛋白的结合能力,在cdk6/cycd3及cdk4/cycd3上进行激酶活性的测定,实验结果如下表5和表6。
[0193]
表5化合物的体外抗肿瘤活性ic
50
(μmol/l)
[0194][0195]
表6化合物分子在200nm和20nm对cdk4及cdk6的抑制率(%)
[0196][0197]
根据上述实验结果我们可以发现,当化合物c-13中的α-氯代酰胺基团替换为乙酰胺基团或丙酰胺基团时,化合物d-1与d-2对三种人乳腺癌细胞株及一种人肺癌细胞株的抑制活性显著下降,较化合物c-13活性降低25倍以上,与palbociclib相比,细胞抑制活性也下降了6倍以上。从表6来看,在浓度为20nm为时,化合物d-1及d-2对cdk6蛋白的抑制率小于50%,基本无抑制剂作用,对cdk4蛋白的抑制率不足70%,远小于还原前的化合物c-13的抑制率81.6%,这也暗示了化合物d-1和d-2与cdk6蛋白基本无结合能力。仅将化合物c-13的α-氯代酰胺基团的cl替换为h或-ch3,不改变分子与cdk6蛋白结合位点的结合模式,同时链长几乎不发生改变,而cl原子替换前后分子活性产生了巨大差异足以表明α-氯代酰胺基团与苏氨酸thr107的共价结合作用在分子活性上发挥了至关重要的作用。
[0198]
实施例5化合物c-13对正常细胞的毒性
[0199]
为了测试目标共价化合物c-13对正常细胞的毒性,我们测试了c-13在浓度为20、10、5、2.5、1.25μm时对人正常肝细胞lo2的抑制率,具体实验方法同实施例2。其结果见图2。
[0200]
从图2中可以看出,当给药浓度为20μm时,化合物c-13对人正常肝细胞lo2肝细胞的抑制率为31.03%,较palbociclib下降30%;在浓度为10μm时,palbociclib的抑制率几乎为c-13的两倍,当浓度继续降低,两者之间的抑制差异不大。目前,临床上关于阳性药palbociclib的毒性报道较少,鉴于图上结果,我们认为化合物c-13对正常细胞具有较低的毒性,处于可接受范围之内,也暗示该共价结合模式不会导致化合物毒性增强。
[0201]
实施例6化合物c-13的细胞周期实验
[0202]
1、实验原理
[0203]
细胞分为处于静止期(g0)和分裂状态的细胞,分裂状态的细胞分为g1期,s期,g2期和m期,细胞内的遗传物质会随着细胞周期进程而发生变化。我们采用pi(即碘化丙啶)标记的方法,它能与细胞内的dna和rna结合,先用rnase酶将rna消化,然后通过流式细胞仪检测dna与pi结合所产生的的荧光强度来直接反映细胞内的dna含量。
[0204]
2、试剂与材料
[0205]
mda-mb-231(人乳腺癌细胞),含有10%胎牛血清的dmem完全培养基,pi染色细胞周期检测试剂盒(凯基),co2培养箱,rnase a酶,0.25%胰酶,pbs(4℃预冷),70%乙醇,碘化丙啶染色液,冷冻离心机,高倍显微镜,流式细胞仪。
[0206]
3、实验步骤
[0207]
取对数生长期的mda-mb-231肿瘤细胞用0.25%胰酶消化,离心,接着用含10%胎牛血清的dmem完全培养基稀释,以1
×
106/孔的细胞密度接种于6孔板。接种完成后,将6孔板置于co2孵箱,37℃、5%co2条件下培养24小时。待mda-mb-231细胞贴壁生长到一定密度后给药,将预先用dmso配制目标化合物c-13溶液用培养基倍比稀释,设置给药浓度为1.5、
0.75、0.375、0.1875、0μm,同时设立阳性对照palbociclib浓度为1.5、0.75、0.375、0.1875、0μm,每个给药浓度均为3个复孔。给药后,co2培养箱孵育12小时,用胰酶消化细胞,将细胞悬液收集到10ml离心管,离心5分钟。吸出上清液,加入1ml预冷的pbs重悬细胞,再次离心沉淀5分钟,重复此步骤洗涤,离心。然后缓慢加入750μl预冷的70%乙醇吹打混匀后4℃固定两小时。去除乙醇,离心5分钟,用pbs重复洗涤三次。加入200μl预冷的pbs重悬细胞,加入rnase a酶20μl,37℃水浴30min。然后每管样品加入400μlpi染色液,4℃下作用30分钟后用流式细胞仪检测。
[0208]
4、结果分析讨论
[0209]
化合物c-13及阳性对照药物palbociclib作用于mda-mb-231人乳腺癌细胞的流式细胞周期分布图及定量数据如图3和图4。
[0210]
从图上数据我们可以分析发现,未加药的空白对照组(control)细胞周期分布为:g1期53.18%,s期23.71%,g2期21.11%,而化合物c-13给药12小时后细胞周期各阶段状态发生不同程度的变化。g1期细胞占比随着化合物c-13化合物给药浓度的增大而不断增加,并呈现浓度依赖性,当浓度为1.5μm时,此时g1期细胞比例为71.12%,较空白对照组提高18%,呈现明显的g1期阻滞。与此同时,s期的细胞比率也从空白对照组的23.71%下降到7.55%。可见,化合物c-13能够通过对cdk6蛋白的共价抑制作用,导致肿瘤细胞dna复制受阻,阻滞细胞周期于g1期。
[0211]
实施例7化合物c-13的细胞凋亡分析
[0212]
1、实验原理
[0213]
该实验方法通过annexin v-fitc/pi双染色法检测肿瘤细胞凋亡活性。磷脂酰丝氨酸(ps)正常情况下只分布于细胞膜脂质双分子层的内侧,当细胞处于凋亡早期,位于细胞膜内侧的磷脂酰丝氨酸翻向外侧。annexin v是一种钙离子依赖性磷脂结合蛋白,它对ps有高度亲和力,因此可通过暴露于细胞膜外侧的磷脂酸丝氨酸与凋亡早期细胞的胞膜结合。annexin v也被称为检测细胞早期凋亡的重要指标。但是,ps由膜内侧翻向外侧这一现象也存在于坏死细胞中,坏死细胞区别于早期凋亡细胞的特征为坏死细胞膜的完整性在早期就已经被破坏。碘化丙啶(pi)染液不能透过完整细胞膜,难以进入凋亡早期细胞及正常细胞内,却能进入死细胞与凋亡中晚期细胞中,结合细胞核呈现红色。因此将annexin v与pi联合使用,可以达到将不同凋亡时期的细胞区分开来的目的。经过染色处理后,采用流式细胞仪检测分析得到细胞凋亡率。
[0214]
2、试剂与材料
[0215]
mda-mb-231(人乳腺癌细胞),6孔板,含有10%胎牛血清的dmem完全培养基,细胞凋亡检测试剂盒(凯基),co2培养箱,0.25%胰酶,pbs(4℃预冷),碘化丙啶染色液,冷冻离心机,高倍显微镜,流式细胞仪。
[0216]
3、实验步骤
[0217]
将对数生长期的mda-mb-231肿瘤细胞用0.25%胰酶消化后,用含10%胎牛血清的dmem完全培养基稀释,以5
×
104/孔的细胞密度接种于6孔板。接种完成后,将6孔板置于co2孵箱,37℃、5%co2条件下培养24小时。待mda-mb-231细胞贴壁生长到一定密度后给药,将预先用dmso配制目标化合物。
[0218]
c-13溶液用培养基倍比稀释,设置给药浓度为0、0.625、1.25、2.5、5、10μm。给药
后,co2培养箱孵育12小时,用0.25%胰酶消化细胞,将单细胞悬液转移到离心管,离心5分钟。吸出上清液,用预冷的pbs缓冲液洗涤,离心,重复此操作两次。然后加入195μl annexin v-fitc重悬细胞,加入5μl annexin v-fitc混匀,25℃下避光孵育10分钟,再补加10μl pi染色液混匀后上机进行检测。annexin v-fitc为绿色荧光,pi为红色荧光。
[0219]
4、结果分析讨论
[0220]
经流式细胞仪测定化合物c-13对于mda-mb-231细胞凋亡情况如图5所示。
[0221]
图5的二维散点图中,十字架将图片分为四个象限。annexin v染色分为左右两部分,右上/右下象限为annexin v染色阳性;pi染色分为上下两部分,左上/右上象限为pi染色阳性。左下象限为双阴结果(annexin v-fitc)-/pi-,此区域为活细胞;左上象限为(annexin v-fitc)-/pi+,此区域包含坏死细胞及少数晚期凋亡细胞;右上象限为双阳结果(annexin v-fitc)+/pi+,此区域为晚期凋亡细胞;右下象限为(annexin v-fitc)+/pi-,此区域代表早期凋亡细胞。
[0222]
从图上数据我们可以明显看出:空白对照组细胞凋亡率为2.67%,化合物c-13的早期凋亡率较空白组(control)明显增加,并且随着给药浓度的增加而不断增加,当浓度为10μm时,肿瘤细胞早期凋亡率升至68.48%。因此,我们可以推测化合物c-13能诱导人乳腺癌细胞mda-mb-231发生凋亡,凋亡能力较强且具有浓度依赖性。
[0223]
实施例8化合物c-13的免疫印迹实验
[0224]
1、实验原理
[0225]
免疫印迹实验又称western blot,是一种常用的蛋白质分析、分离技术。采用的是聚丙烯酰胺凝胶电泳技术,在电场的作用下将page分离的蛋白质转移到固相载体,固相支持物在吸附蛋白质的同时不改变其空间构象及生物活性。然后以固定相上的蛋白质作为抗原,利用抗原-抗体高度特异性结合的原理,检测出蛋白质混合物中的目标蛋白,从而定性或定量的分析细胞或组织中目标蛋白的表达情况。
[0226]
2、试剂与材料
[0227]
人乳腺癌细胞mda-mb-231,含有10%胎牛血清的dmem完全培养基,0.25%胰酶,co2培养箱,pbs缓冲液(4℃预冷),bca蛋白浓度测定试剂盒,cdk4(d9g3e)rabbit mab,cdk6(d4s8s)rabbit mab,phospho-rb(ser780)(d59b7)rabbit mab,兔igg抗体(a7016),ripa裂解液,sds-page试剂,匀浆缓冲液,转膜缓冲液,膜染色液,显色液等。
[0228]
3、具体步骤
[0229]
细胞培养及给药:将处于对数生长期的肿瘤细胞用0.25%胰酶消化,离心,接着用含10%胎牛血清的dmem完全培养基稀释,以合适的细胞密度接种于6孔板,每孔培养基1ml。接种完成后,将6孔板置于co2孵箱,37℃、5%co2条件下培养24小时。待肿瘤细胞贴壁生长到一定密度后给药,将提前配制的目标化合物c-13溶液用培养基倍比稀释,设置给药浓度为40、20、10、5、2.5、0μm,阳性对照药物palbociclib给药浓度为20、10μm。给药后培养24小时,收集细胞,用提前预冷的pbs缓冲液洗涤,吸出上清液,离心,重复三次。
[0230]
总蛋白的提取:加入适当体积的ripa裂解液,轻轻摇匀后置于冰上裂解30分钟,并经常摇动。裂解完成后,将细胞刮于6孔板的一侧,在冰上转移至1.5ml离心管,在提前预冷的离心机上12000rpm离心五分钟,吸出上清液移至0.5ml离心管置于-20℃下保存。
[0231]
sds-page电泳:选用聚丙烯酰胺凝胶分离蛋白质。
[0232]
上样:将配制好的胶板置于电泳槽,加足够的电泳液后开始上样,使用微量进样器针头插入加样孔缓慢加入样品。
[0233]
电泳:在40v电压下电泳4-5小时,至溴酚兰刚跑出时终止电泳。
[0234]
转膜:将玻璃板的凝胶取出,根据实验所需截取合适的凝胶,然后放置于转膜缓冲液中。将大小适宜的pvdf膜放入转膜装置,按顺序叠放好后,夹紧,置于转移槽,加满转膜缓冲液。把整个装置放入冰浴中,开始通电转膜。转膜45分钟后,取出pvdf膜,置于tbs缓冲液。
[0235]
免疫反应:将膜用tbs浸湿后移至含封闭液的平皿中,摇床上摇动封闭一小时。用tbst稀释一抗,将抗体溶液加到保鲜膜上,取出封闭液中的pvdf膜,将膜蛋白面朝下放在抗体液面上,赶走气泡,室温下孵育两小时,用tbst摇床上洗涤两次,再用tbs缓冲液洗涤一次。二抗的操作方法同此步骤。
[0236]
发光反应及显影:按照试剂盒说明配备显影液,将pvdf膜浸入溶液中,一段时间后上机检测。然后对条带进行分析,使用actin作为内参。
[0237]
4、结果分析讨论
[0238]
发光反应上机检测后,经x胶片曝光显影,所得条带结果如图6所示。
[0239]
为了进一步研究化合物c-13对cyclin d-cdk4/6-rb通路的影响,进行了western blot实验。观察图7的结果,我们可以发现化合物c-13作用于mda-mb-231细胞24h后能有效抑制剂prb(ser780)的表达,且对其抑制能力呈现浓度依赖性。这表明化合物c-13能够抑制rb蛋白的磷酸化作用,从而阻断cdk4/6
→
rb
→
e2f信号通路,与cdk4/6抑制剂能够与cyclin d-cdk6复合物结合,阻止rb磷酸化及转录因子e2f释放的作用机制相吻合。
[0240]
实施例9化合物c-13的平板克隆实验
[0241]
1、实验原理
[0242]
平板克隆又称作克隆形成实验,是测定细胞增殖能力的有效方法之一。细胞贴壁后不一定都能增殖和形成克隆,但形成克隆的细胞必定为有增殖活力的贴壁细胞。我们用细胞克隆形成率来表示贴壁细胞增殖并形成克隆的数量,即细胞接种存活率。通过结晶紫染色直观的从颜色上判断克隆的形成,反映药物作用下肿瘤细胞的抗增殖能力。
[0243]
2、试剂与材料
[0244]
人乳腺癌细胞mda-mb-231,含有10%胎牛血清的dmem完全培养基,0.25%胰酶,co2培养箱,pbs缓冲液(4℃预冷),0.5%结晶紫染色液,4%多聚甲醛固定液,6孔板。
[0245]
3、具体步骤
[0246]
取对数生长期的mda-mb-231肿瘤细胞用0.25%胰酶消化并吹打成单个悬浮细胞后,用含10%胎牛血清的dmem完全培养基稀释,以1
×
103/孔的细胞密度接种于6孔板。将6孔板置于37℃、5%co2细胞培养箱中培养24小时。待mda-mb-231细胞贴壁生长到一定密度后给药,将提前配备的目标化合物c-13溶液用培养基倍比稀释,设置给药浓度为0、0.1563、0.3125、0.625、1.25μm。加药后,放置于co2培养箱中培养14天。当形成肉眼可见的克隆后,停止培养。然后吸出上清液,pbs缓冲液洗涤两次。加入4%多聚甲醛固定液5ml,固定15分钟。弃去固定液,加适量0.5%结晶紫染色液染色20分钟,洗去染液,空气下干燥。照相机拍照取图,分析克隆形成。
[0247]
4、结果分析讨论
[0248]
培养14天后,化合物c-13作用于mda-mb-231所致的克隆形成如图7所示。
[0249]
由实验数据我们可以看出:化合物c-13对人乳腺癌细胞mda-mb-231产生明显的克隆形成抑制作用。与未给药的空白对照组control相比,给药后的细胞克隆形成数明显降低,且随药物浓度增大而减少。当药物浓度为0.625μm时,已基本无明显的克隆集落形成。因此可得知化合物c-13能有效抑制mda-mb-231肿瘤细胞的增殖,与mtt实验结果相吻合。
[0250]
实施例10化合物c-13(c13)的体内抗肿瘤实验
[0251]
为了研究c13在体内抗肿瘤作用,我们综合c13在体外活性的数据,选取了体外活性较好的mda-mb-231作为实验细胞株作为体内模型造瘤株。本实验动物为balb/c nude小鼠。
[0252]
实验动物饲养与要求:本课题组实验所用的裸鼠均为北京华阜康生物科技股份有限公司提供,于四川大学生物治疗国家重点实验室(spf级)动物房饲养,所购买的裸鼠均为spf级。所有动物实验由四川大学实验动物管理委员会的批准下进行,饲养规则按national institutes of health guide for the care and use of laboratory animals国际标准执行。实验动物饲养条件:温度20℃,相对湿度35%-60%,采用人工照明系统,12h明暗交替,co60灭菌饲料(北京科澳协力饲料有限公司),灭菌饮水,每周定期更换两次笼子和垫料(均灭菌),保障小鼠生活空间为干燥洁净的环境。
[0253]
动物模型的建立:收集对数期的mda-mb-231细胞(1
×
107)经皮下注射到6-7周龄的雌性balb/c小鼠中。当接种后裸鼠平均肿瘤体积增长到大约100mm3时,将小鼠随机分为四组(每组n=3):对照组:(10%dmso,10%peg-300和50mm乳酸钠溶液),化合物c13(40mg/kg,80mg/kg溶于10%dmso,10%peg-300和50mm乳酸钠溶液中),palbociclib(40mg/kg溶于10%dmso,10%peg-300和50mm乳酸钠溶液),通过经口灌胃的方式给药。每三天测定一次肿瘤大小和体重。
[0254]
用游标卡尺测量肿瘤体积,具体见图8,肿瘤体积的计算公式为:0.5
×
最短直径2×
最长直径。肿瘤生长的抑制率的公式为:100
×
{1-[最终肿瘤体积
实验组-肿瘤初始体积
实验组
]/[最终肿瘤体积
空白组-肿瘤初始体积
空白组
]}。实验时间为21天,实验结束后,用水合氯醛处死小鼠,剥离肿瘤,心、肝、脾、肺、肾等脏器。将肿瘤进行称重后一并分组与脏器保存于4%多聚甲醛。试验结果见图9和图10。
[0255]
根据实验结果,c13在40mg/kg和80mg/kg给药浓度下,对肿瘤的抑制率(抑瘤率)分别为93.49%和104.68%,,阳性药palbociclib在40mg/kg给药浓度下抑瘤率为86.82%。可见,同等浓度下,c13的体内抗肿瘤活性高于阳性药palbociclib。且在给药过程中,c13给药组的小鼠体重平稳,显示了该分子的低毒性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1