一种制备地瑞那韦中间体的方法与流程
本申请是申请号201610299132.8,申请日2016年5月7日,发明创造名为“一种制备地瑞那韦中间体的方法”的分案申请。
本发明属于医药技术领域,具体涉及艾滋病药物地瑞那韦中间体的制备方法。
背景技术:
地瑞那韦(darunavir)是美国强生制药公司开发的一种用于治疗艾滋病的药物,地瑞那韦乙醇合物(darunavirethanolate)于2006年首次在美国和加拿大上市。
地瑞那韦英文名称:
[(3r,3as,6ar)-2,3,3a,4,5,6a-hexahydrofuro[5,4-b]furan-3-yl]n-[(2s,3r)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate
分子式:c27h37n3o7s
分子量:547.66
地瑞那韦是一种新的用于艾滋病治疗的非肽类抗逆转病毒蛋白酶抑制剂,由强生制药冰岛分公司tibotec首次研发成功,是目前6种蛋白酶抑制剂(沙奎那韦,利托那韦,茚地那韦,萘非那韦,安瑞那韦及abt378/r)中生物利用度最高的,通过阻断从受感染的宿主细胞表面释放新的、成熟的病毒粒子的形成过程,抑制病毒的蛋白酶而起作用。当长期应用本品时,通常可降低血中的hiv病毒载体,增加cd4细胞的计数,降低感染艾滋病的机会,提高生活质量,延长生命。适用于感染了艾滋病病毒但服用现有抗逆转录病毒药物未见疗效的成年人,该药必须与低剂量的利托那韦或其他抗逆转病毒制剂结合使用,以提高药效。通过对抗急性及慢性受感染的成淋巴细胞以及外周系统血中的淋巴细胞可评价其体外抗病毒活性,其ic50对急性感染细胞是0.012~0.08mmol/l,对慢性感染细胞是0.41mmol/l。口服用药,推荐剂量1次1200mg,1日2次,轻至中度肝功能不良的患者及肾功能不良者应减量。地瑞那韦的不良反应主要为胃肠道反应、潮红、瘙痒及口周麻木感、抑郁、情绪紊乱、味觉紊乱等。中至重度肝功能不良的患者不推荐使用本品。由于本组分中含有磺胺基团,因此对磺胺过敏者及对本品处方中任一组分过敏者禁用。
地瑞那韦乙醇合物(darunavirethanolate)于2006年首次在美国和加拿大上市,商品名为prezista,之后陆续在澳大利亚、日本等多个国家上市。地瑞那韦乙醇合物与cobicistat的复方prezcobix也于2014年上市。
剂型:口服悬浮液,100mg/ml;片剂,75mg,150mg,600mg,800mg。
用法用量:一般患者,800mg地瑞那韦乙醇合物和100mg利托那韦与食物同时服用,一日一次。对地瑞那韦有抵触的患者,600mg地瑞那韦乙醇合物和100mg利托那韦与食物同时服用,一日两次。儿童用量不得超过成人用量,具体遵医嘱。
地瑞那韦销售:
2012年14.14亿美元;2013年16.73亿美元;2014年18.31亿美元(含prezcobix复方);2015年前三季度13.21亿美元(含prezcobix复方)。
(3r,3as,6ar)-羟基六氢呋喃并[2,3-b]呋喃-3-醇为地瑞那韦的关键中间体。
文献jmedchem,1996,39:3278-3290报道了以d-苹果酸二乙酯为起始原料合成该中间体的方法以及以二氢呋喃为原料经过酶拆分合成此中间的方法,反应路线如下所示:
该方法原料成本高,部分反应条件要求较高不利于放大生产和控制。
此后,文献orgprocessres.dev.,2007,11:972-980和org.lett.,2008,10:1103-1106分别报道了以二氢呋喃和二聚羟基乙醛为原料合成该中间体相关的方法,反应路线如下所示:
此方法手性诱导效果不理想,需要进一步用酶拆分的方法才能获得光学纯的产品。
专利wo03022853a1公开了更为可靠的合成方法,此方法以价格低廉的以异vc为起始原料经过多步反应合成地瑞那韦中间体,反应路线如下所示:
但是该合成方法原子经济性差,由于需要用到价格较高的高碘酸和硼氢化锂,导致产品的原料成本较高。
技术实现要素:
本发明针对现有技术制备地瑞那韦中间体(3r,3as,6ar)-羟基六氢呋喃并[2,3-b]呋喃-3-醇的缺陷,公开了一种简洁、经济的制备方法。
具体发明内容如下:
1.本发明涉及一种式i化合物的制备方法,包括以下步骤:
(1)式iii化合物制备得到化合物ii;
(2)式ii化合物制备得到化合物i。
2.如1所述的方法,其中步骤(1)在催化剂存在条件下进行不对称催化反应,制备得到化合物ii,其中所述的催化剂为[(s)-binap]rucl2、[(s)-binap]ru(oac)2、[(s)-segphos]ru(oac)2或(s)-ru(h8-binap)(oac)2中的一种或一种以上的混合催化剂。
3.如1所述的方法,其中步骤(2)在还原剂存在的条件下制备得到化合物i,其中所述的还原剂为红铝、lialh(otbu)3、dibal-h中的一种或一种以上的混合还原剂。
4.如1所述的方法,其中式iii化合物可以按照下述方法制备,
式iv化合物和1,4-丁内酯在碱存在下,反应制备得到式iii化合物,其中所述的碱为正丁基锂、二异丙基氨锂、双(三甲基硅基)氨基锂(lhmds)中的一种或一种以上的混合碱。式iv化合物可以参照文献j.org.chem.1993,58,7768中报道的方法制备。
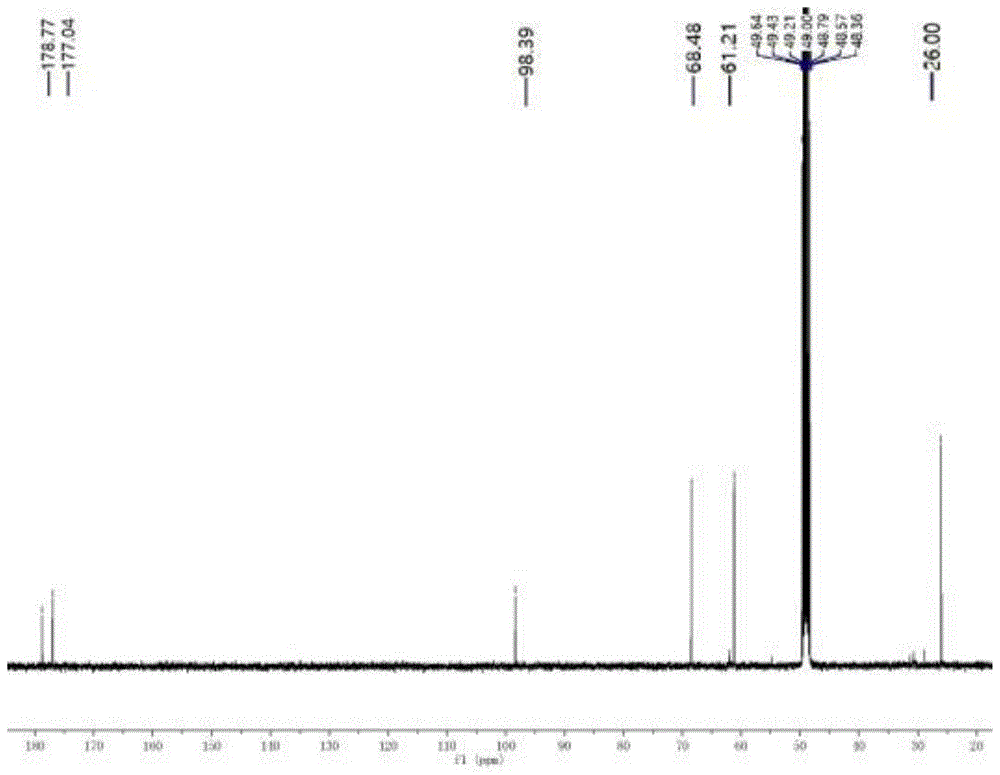
5.本发明还涉及式ii,式iii所示化合物,式iii化合物的核磁图谱及质谱见说明书附图(图1、图2、图3)。
附图说明
图1为式iii化合物的核磁c谱示意图;
图2为式iii化合物的核磁h谱示意图;
图3为式iii化合物的质谱图。
实施例
通过下列非限定性实施例对本发明进一步举例说明。
式iii化合物的合成
实施例一
氮气保护下,向500ml烧瓶中加入270ml1.6mol/l的正丁基锂的正己烷溶液,冷却到-78℃,滴加34.4g(2.0eq)1,4-丁内酯。滴加完毕后,保温搅拌2小时,然后滴加31.2g(1.0eq,0.2mol)1,4-二氧杂螺[4,5]癸-2-酮(式iv化合物)的四氢呋喃溶液。保温反应16h后,用饱和氯化铵溶液淬灭反应,升温到室温,分层,水相用乙酸乙酯萃取,合并有机相,浓缩后得产品,收率38%。
1hnmr(cd3od,400mhz):δ4.64(s,2h),3.72(t,j=6.3hz,2h),2.42(t,j=6.3hz,2h)
13cnmr(cd3od,400mhz):δ178.77,177.02,98.39,68.48,61.21,26.00
hrms:m-1=143.0352
熔点:115-117℃。
实施例二
氮气保护下,向250ml烧瓶中加入160ml2.0mol/l的二异丙基氨锂溶液,冷却到-78℃,滴加21.5g(2.5eq)1,4-丁内酯。滴加完毕后,保温搅拌2小时,然后滴加15.6g(1.0eq,0.1mol)1,4-二氧杂螺[4,5]癸-2-酮(式iv化合物)的四氢呋喃溶液。保温反应8h后,用饱和氯化铵溶液淬灭反应,升温到室温,分层,水相用乙酸乙酯萃取,合并有机相,浓缩后得产品,收率40%。
实施例三
氮气保护下,向250ml烧瓶中加入220ml1.0mol/l的双(三甲基硅基)氨基锂(lhmds)溶液,冷却到-78℃,滴加17.2g(2.0eq)1,4-丁内酯。滴加完毕后,保温搅拌2小时,然后滴加15.6g(1.0eq,0.1mol)1,4-二氧杂螺[4,5]癸-2-酮(式iv化合物)的四氢呋喃溶液。保温反应12h后,用饱和氯化铵溶液淬灭反应,升温到室温,分层,水相用乙酸乙酯萃取,合并有机相,浓缩后得产品,收率35%。
式ii化合物的合成
实施例一
将5.04g(0.035mol)化合物iii,加入到100ml高压釜中,加入50ml甲醇,搅拌溶解。用高纯氮气转换釜内空气三次,然后在氮气保护下,向釜内加入1.4g(0.05eq)[(s)-binap]rucl2,用高纯氢气置换釜内空气三次,充氢气至6.0mpa,加热至80-90℃反应24小时。冷却至室温,小心排放釜内空气,取出反应液,浓缩后得化合物ii,产物ii未经分离,直接进入下一步反应。
实施例二
将5.76g(0.04mol)化合物iii,加入到100ml高压釜中,加入60ml乙醇,搅拌溶解。用高纯氮气转换釜内空气三次,然后在氮气保护下,向釜内加入0.17g(0.005eq)[(s)-binap]ru(oac)2,用高纯氢气置换釜内空气三次,充氢气至6.0mpa,加热至60-70℃反应60小时。冷却至室温,小心排放釜内空气,取出反应液,浓缩后得化合物ii,产物ii未经分离,直接进入下一步反应。
实施例三
将5.76g(0.04mol)化合物iii,加入到100ml高压釜中,加入60ml乙酸乙酯,搅拌溶解。用高纯氮气转换釜内空气三次,然后在氮气保护下,向釜内加入0.34g(0.01eq)[(s)-segphos]ru(oac)2,用高纯氢气置换釜内空气三次,充氢气至4.0mpa,加热至50-60℃反应72小时。冷却至室温,小心排放釜内空气,取出反应液,浓缩后得化合物ii,产物ii未经分离,直接进入下一步反应。
实施例四
将5.76g(0.04mol)化合物iii,加入到100ml高压釜中,加入60ml甲醇,搅拌溶解。用高纯氮气转换釜内空气三次,然后在氮气保护下,向釜内加入0.17g(0.005eq)(s)-ru(h8-binap)(oac)2,用高纯氢气置换釜内空气三次,充氢气至2.5mpa,加热至30-40℃反应72小时。冷却至室温,小心排放釜内空气,取出反应液,浓缩后得化合物ii,产物ii未经分离,直接进入下一步反应。
式i化合物的合成:
实施例一
氮气保护下,向500ml四口瓶中加入120.0g(4.47eq)68%红铝溶液,再加入228.0g甲苯,冷却到-10~-20℃后滴加40.5g(1.49eq)三氟乙醇,滴加完毕后升温至20-30℃反应1-2小时。加入含有13.1g(1.00eq)化合物ii的100ml四氢呋喃溶液,控制温度在-10~0℃反应至原料ii消失。控制温度在-20-15℃,将反应液加入到200.0g20%的硫酸中,搅拌20h后静置分层,收集有机相,水相用100g甲苯萃取,合并甲苯层,用5%碳酸氢钠洗涤,再用水洗涤,有机相减压浓缩,得产品29.5g,收率84.5%。
实施例二
氮气保护下,向四口瓶中加入14g(1.00eq)化合物ii和的300ml甲苯,搅拌,冷却至-20~-15℃,分批次加76.5g(3.15eq)lialh(otbu)3,保温反应到原料ii消失。将反应液加入到120.0g16%的盐酸中,升温到30℃搅拌反应30小时后静置分层,收集有机相,水相用100g甲苯萃取,合并甲苯层,用5%碳酸氢钠洗涤,再用水洗涤,有机相减压浓缩,得粗产品33.1g,94.6%。
实施例三
氮气保护下,向四口瓶中加入24.3g(1.00eq)化合物ii和的260ml甲苯,搅拌,冷却至-50~-40℃,滴加315g(3.3eq)dibal-h的25%甲苯溶液,保温反应到原料ii消失。将反应液加入到200.0g16%的盐酸中,加热到60℃反应16小时,静置分层,收集有机相,水相用150g甲苯萃取,合并甲苯层,用5%碳酸氢钠洗涤,再用水洗涤,静置后分层,有机相减压浓缩,得产品61.7g,收率95%。
- 还没有人留言评论。精彩留言会获得点赞!