一种喹啉生物碱类化合物及其制备方法与应用与流程
1.本发明属于植物化学技术领域,具体涉及一种喹啉生物碱类化合物及其制备方法与应用。
背景技术:
2.烟草是茄科烟草属植物,是世界上种植最广泛的经济作物之一。而中国是世界上最大的烟草生产国和消费国,年产量可达4~5亿吨。在烟草种植和卷烟生产过程中,每年会产生超过2亿吨的废弃烟草材料,如烟草的根、茎、低等级或有缺陷的烟叶等,如何加强烟草及其废弃物的综合利用引起了科研工作者的广泛关注。
3.根据文献报道,目前从烟草中鉴定出的化合物高达4000多种,主要成分包括二萜类化合物、倍半萜类化合物、黄酮类化合物、生物碱、香豆素等。同时有研究证明,上述化合物具有不同的药理学作用,如抗菌、抗氧化、抗肿瘤、抗烟草花叶病毒等。因此,提高烟草及其废弃物的药用价值和副产品的综合利用具有十分重要的意义。
技术实现要素:
4.本发明从烟草茎中分离得到了一种具有抗烟草花叶病毒活性的新生物碱类化合物,该化合物至今尚未见到相关报道。
5.除非另有说明,本发明中所采用的百分数均为质量百分数。
6.本发明第一方面提供一种喹啉生物碱类化合物,所述化合物分子式为:
7.c
15
h
17
no2,其具有下述结构:
[0008][0009]
该化合物为棕色胶状物,命名为:4
‑
乙酰
‑7‑
异丙基
‑6‑
甲基喹啉
‑
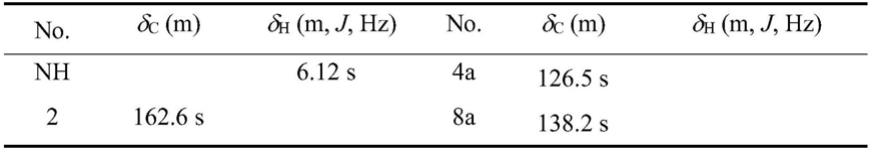
2(1h)
‑
酮,英文名为:4
‑
acetyl
‑7‑
isopropyl
‑6‑
methylquinolin
‑
2(1h)
‑
one。
[0010]
本发明第二方面提供第一方面所述的喹啉生物碱类化合物的制备方法,具体包括以下步骤:
[0011]
a、浸膏提取:取烟草茎为原料,粉碎,用90wt%~99wt%乙醇超声提取后,提取液加入到乙酸乙酯与3wt%的酒石酸混合液中,待混合溶液静置分层后,再用na2co3溶液将水层ph值调节至9.0,并用乙酸乙酯再次萃取,经过滤,减压浓缩成浸膏;
[0012]
b、硅胶柱层析:将步骤a得到的浸膏用200~300目硅胶干法装柱,进行硅胶柱层析;以体积配比分别为10:0、9:1、8:2、7:3、6:4、5:5的氯仿
‑
甲醇溶液进行梯度洗脱,合并相同极性的部分,收集各部分洗脱液并浓缩;其中,所述硅胶与所述浸膏的质量比为2~5;收
集其中用体积比为9:1的氯仿
‑
甲醇溶液洗脱时得到的洗脱液,称为第一洗脱液;将上述第一洗脱液用硅胶层析柱继续分离,用体积配比依次为9:1、8:2、7:3、6:4、5:5的一系列氯仿
‑
丙酮溶液进行梯度洗脱,收集其中用体积比为8:2的氯仿
‑
丙酮溶液洗脱时得到的洗脱液,称为第二洗脱液;
[0013]
c、高效液相色谱分离:将b步骤最终得到的第二洗脱液通入高效液相色谱进行分离纯化,所述高效液相色谱法分离纯化是采用21.2mm
×
250mm,5μm的zorbaxprepht gf色谱柱,流速为20ml/min,流动相为53wt%的甲醇水溶液,紫外检测器检测波长为338nm,第三洗脱液每次进样200μl,收集每次进样后色谱峰保留时间为18.6min时所对应的洗脱液,称为第三洗脱液,将该第三洗脱液脱除溶剂后即得所述的喹啉生物碱类化合物。
[0014]
优选地,所述步骤a中,乙醇的浓度优选为95wt%。
[0015]
优选地,步骤b中,所述浸膏在经硅胶柱层析粗分前,用甲醇溶解后用重量比1.5~2.5倍的80~120目硅胶拌样。
[0016]
优选地,所述步骤c中,高效液相色谱法分离纯化后,所得化合物再次用纯甲醇溶解,再以纯甲醇为流动相,用凝胶柱层析分离,以进一步分离纯化。
[0017]
本发明第三方面提供第一方面所述的喹啉生物碱类化合物在制备抗烟草花叶病毒药物中的应用。
[0018]
以上述方法制备的喹啉生物碱类化合物的结构是通过以下方法测定出来的:
[0019]
外观观测发现:本发明化合物为棕色胶状物;
[0020]
紫外
‑
可见吸收光谱显示其在232与338nm处有最大吸收,证明该化合物中存在芳香环结构;
[0021]
红外光谱(溴化钾压片)显示该化合物中有胺基(3281cm
‑1)、羰基(1677、1650cm
‑1)、芳香环(1614和1455cm
‑1)特征官能团;高分辨质谱(hresims)给出准分子离子峰266.1150[m+na]
+
,可确定化合物的分子式为c
15
h
17
no2,不饱和度为8。结合1h和
13
c与hsqc nmr数据,显示该化合物包括一个1,2,4,5
‑
四取代的苯环(c
‑
5~c
‑
8,c
‑
4a和c
‑
8a,h
‑
5和h
‑
8),一个异丙基(c
‑
4'~6',h
‑
4',和h6
‑
5',6'),一个乙酰基(c
‑
1',c
‑
2',和h3‑
2'),一个
‑
nh
‑
co
‑
ch
‑
c
‑
基团(c
‑
2~4,h
‑
1,h
‑
3和nh)。为了满足化合物的8个不饱和度,两个羰基、一个双键、苯环和
‑
nh
‑
co
‑
ch
‑
c
‑
基团应形成1个喹啉
‑
2(1h)
‑
酮结构。该推断可进一步由h
‑
3与c
‑
2、c
‑
4、c
‑
4a,nh与c
‑
2、c
‑
3、c
‑
8、c
‑
4a、c
‑
8a,h
‑
8与c
‑
4a、c
‑
8a,h
‑
5与c
‑
4、c
‑
4a、c
‑
8a的hmbc相关得到证实。
[0022]
化合物的母体骨架确定后,剩余的取代基位置可通过进一步分析其hmbc相关确定。根据h
‑
2'与c
‑
4,h
‑
3与c
‑
1'的hmbc相关,可确定乙酰基取代在c
‑
4位;根据h
‑
4'与c
‑
6、c
‑
7、c
‑
8,h6
‑
5',6'与c
‑
7的hmbc相关,可确定异丙基取代在c
‑
7位。最后,根据h3‑
3'与c
‑
5、c
‑
6、c
‑
7的hmbc相关,可确定甲基(c
‑
1')取代在c
‑
6位。至此,本发明化合物的结构得到确认,鉴定为4
‑
乙酰
‑7‑
异丙基
‑6‑
甲基喹啉
‑
2(1h)
‑
酮。
[0023]
表
‑
1.化合物的1hnmr和
13
c nmr数据(cdcl3)
[0024]
[0025][0026]
本发明的有益效果如下:
[0027]
1、本发明首次从烟草茎中分离出一种新的化合物,通过核磁共振和质谱测定方法确定了为喹啉生物碱类化合物,并表征了其具体结构。且该喹啉生物碱类化合物具有很好的抗烟草花叶病毒活性:经对抗烟草花叶病毒的实验,发现该喹啉生物碱类化合物的相对抑制率达到38.5%,其活性高于对照品宁南霉素的相对抑制率(33.0%)。以上结果揭示了本发明的化合物在制备抗烟草花叶病毒药物中有良好的应用前景。本发明化合物结构简单活性好,可作为抗烟草花叶病毒药物的先导性化合物。
[0028]
2、由于烟草根茎生物产量巨大,在烟叶生产过程中一般作为废弃物丢弃,本发明的化合物原料易得;化合物的提取方法简单,容易分离得到,产业化制备容易实现。
[0029]
3、本发明化合物分子结构也简单,容易实现人工合成,后续的产业化也可通过人工合成实现。
[0030]
4、采用了常规柱层析和高效液相色谱结合的制备方法,化合物制备操作流程简单,所获得的本发明化合物纯度高,后续的工业化生产中化合物的品质、纯度有保障。
[0031]
5、本发明化合物安全无毒,展现出良好的抗烟草花叶病毒活性,可为烟草花叶病的防治提供理想的新骨架类型药源分子。
附图说明
[0032]
图1为所述喹啉生物碱类化合物的核磁共振碳谱。
[0033]
图2为所述喹啉生物碱类化合物的核磁共振氢谱。
[0034]
图3为所述喹啉生物碱类化合物的主要hmbc相关。
具体实施方式
[0035]
下面对本发明通过实施例作进一步说明,但不仅限于本实施例。实施例中未注明具体条件的实验方法,通常按照常规条件以及手册中所述的条件,或按照制造厂商所建议的条件所用的通用设备、材料、试剂等,如无特殊说明,均可从商业途径得到。以下实施例中所需要的原料均为市售。
[0036]
本发明所述喹啉生物碱类化合物c
15
h
17
no2的制备方法包括浸膏提取、硅胶柱层析、高压液相色谱分离步骤,具体包括:
[0037]
a、浸膏提取:取风干的云烟
‑
116的茎为原料,粉碎至20~40目,用90~99%乙醇超声提取后,提取液加入到乙酸乙酯与3%的酒石酸混合液中,待混合溶液静置分层后,再用na2co3溶液将水层ph值调节至9.0,并用乙酸乙酯再次萃取,经过滤,减压浓缩成浸膏;
[0038]
b、硅胶柱层析:浸膏用200~300目硅胶干法装柱,进行硅胶柱层析;以体积配比分别为10:0、9:1、8:2、7:3、6:4、5:5的氯仿
‑
甲醇溶液进行梯度洗脱,合并相同极性的部分,收集各部分洗脱液并浓缩;其中,所述硅胶与所述浸膏的质量比为2~5。氯仿
‑
甲醇9:1洗脱液浓缩的部分,以体积配比分别为9:1、8:2、7:3、6:4、5:5的氯仿
‑
丙酮溶液进行洗脱,收集洗脱液的8:2部分浓缩,供高效液相色谱分离纯化。
[0039]
c、高效液相色谱法分离纯化是采用21.2mm
×
250mm,5μm的zorbaxprepht gf色谱柱,流速为20ml/min,流动相为53%的甲醇水溶液,紫外检测器检测波长为338nm,每次进样200μl,收集18.6min的色谱峰,多次累加后蒸干可得化合物粗品。
[0040]
d、所述c步骤中高效液相色谱法分离纯化后,所得化合物粗品再次用纯甲醇溶解,再以甲醇为流动相,用凝胶柱层析分离,以进一步分离纯化,得到所述喹啉生物碱类化合物。
[0041]
本发明所用原料不受地区和品种限制,均可以实现本发明,下面以来源于云南的烟草茎样品对本发明做进一步说明:
[0042]
实施例1
[0043]
烟草根茎样品来源于云南玉溪,品种为云烟
‑
116。将烟草茎取样2.0kg粉碎到30目,用95%的乙醇超声提取3次,每次提取30min,提取液合并,加入到乙酸乙酯与3%的酒石酸混合液(乙酸乙酯:酒石酸=97:3,质量比)中,待混合溶液静置分层后,再用na2co3溶液将水层ph值调节至9.0,并用乙酸乙酯再次萃取,经过滤,减压浓缩成浸膏,得浸膏98.2g。浸膏用纯甲醇溶解后用150g的100目粗硅胶拌样,1.0kg的200目硅胶装柱进行硅胶柱层析,用体积配比为10:0、9:1、8:2、7:3、6:4、5:5的氯仿
‑
甲醇梯度洗脱,tlc监测合并相同的部分,得到6个部分,其中体积配比为9:1的氯仿
‑
甲醇洗脱部分浓缩,再次以体积配比分别为9:1、8:2、7:3、6:4、5:5的氯仿
‑
丙酮溶液进行洗脱,收集洗脱液的8:2部分,用石油醚
‑
丙酮溶液进行洗脱后,再用安捷仑1100制备高效液相色谱分离,以53%的甲醇为流动相,zorbaxprepht gf柱(21.2
×
250mm,5μm)制备柱为固定相,流速为20ml/min,紫外检测器检测波长为338nm,每次进样200μl,收集18.6min的色谱峰,多次累加后蒸干可得化合物粗品;所得产物粗品再次用纯甲醇溶解,再以纯甲醇为流动相,用sephadex lh
‑
20凝胶柱层析分离,即得该新化合物纯品。
[0044]
实施例2
[0045]
烟草根茎样品来源于云南大理,品种为云烟
‑
116。将烟草茎取样3.5kg粉碎到20目,用99%的乙醇超声提取3次,每次提取30min,提取液合并,加入到乙酸乙酯与3%的酒石酸混合液中,待混合溶液静置分层后,再用na2co3溶液将水层ph值调节至9.0,并用乙酸乙酯再次萃取,经过滤,减压浓缩成浸膏,得浸膏178g。浸膏用纯甲醇溶解后用350g的80目粗硅胶拌样,1.5kg的250目硅胶装柱进行硅胶柱层析,用体积配比为10:0、9:1、8:2、7:3、6:4、5:5的氯仿
‑
甲醇梯度洗脱,tlc监测合并相同的部分,得到6个部分。氯仿
‑
甲醇9:1洗脱液浓缩的部分再次进行硅胶柱层析,以体积配比分别为9:1、8:2、7:3、6:4、5:5的氯仿
‑
丙酮溶液进行洗脱,收集洗脱液的8:2部分浓缩,供高效液相色谱制备用。液相色谱制备再用安捷仑1100半制备高效液相色谱分离,以53%的甲醇为流动相,zorbaxprepht gf柱(21.2
×
250mm,5μm)制备柱为固定相,流速为20ml/min,紫外检测器检测波长为338nm,每次进样200l,收集18.6min的色谱峰,多次累加后蒸干可得化合物粗品;所得产物粗品再次用纯甲
醇溶解,再以纯甲醇为流动相,用sephadex lh
‑
20凝胶柱层析分离,即得该新化合物纯品。
[0046]
实施例3
[0047]
烟草根茎样品来源于云南曲靖,品种为云烟
‑
116。将烟草茎取样5.5kg粉碎到40目,用99%的乙醇超声提取3次,每次提取30min,提取液合并,加入到乙酸乙酯与3%的酒石酸混合液中,待混合溶液静置分层后,再用na2co3溶液将水层ph值调节至9.0,并用乙酸乙酯再次萃取,经过滤,减压浓缩成浸膏,得浸膏245g。浸膏用纯甲醇溶解后用615g的120目粗硅胶拌样,1.8kg的300目硅胶装柱进行硅胶柱层析,用体积配比为10:0、9:1、8:2、7:3、6:4、5:5的氯仿
‑
甲醇梯度洗脱,tlc监测合并相同的部分,得到6个部分。氯仿
‑
甲醇9:1洗脱液浓缩的部分再次进行硅胶柱层析,以体积配比分别为9:1、8:2、7:3、6:4、5:5的氯仿
‑
丙酮溶液进行洗脱,收集洗脱液的8:2部分浓缩,供高效液相色谱制备用。液相色谱之内用安捷仑1100半制备高效液相色谱分离,以53wt%的甲醇为流动相,zorbaxprepht gf柱(21.2
×
250mm,5μm)制备柱为固定相,流速为20ml/min,紫外检测器检测波长为338nm,每次进样200l,收集18.6min的色谱峰,多次累加后蒸干可得化合物粗品;所得粗品再次用纯甲醇溶解,再以纯甲醇为流动相,用sephadex lh
‑
20凝胶柱层析分离,即得该新化合物。
[0048]
实施例4
[0049]
取实施例1制备的化合物,为棕色胶状物。
[0050]
测定方法为:用核磁共振,结合其它波谱技术鉴定结构。
[0051]
1)紫外光谱(溶剂为甲醇),λmax(logε)210(4.18)、232(3.56)、338(3.20)nm;
[0052]
2)红外光谱(溴化钾压片)νmax 3281、2924、1677、1650、1614、1455、1226、1150、859、740cm
‑1;
[0053]
3)高分辨质谱(hresims)给出准分子离子峰266.1150[m+na]
+
(计算值266.1157)。结合1h和
13
c nmr谱给出一个分子式c
15
h
17
no2,不饱和度为8。
[0054]
从1h和
13
cnmr谱(数据归属见表
‑
1)信号可以看出:显示该化合物包括一个1,2,4,5
‑
四取代的苯环(c
‑
5~c
‑
8,c
‑
4a和c
‑
8a,h
‑
5和h
‑
8),一个异丙基(c
‑
4'~6',h
‑
4',和h6
‑
5',6'),一个乙酰基(c
‑
1',c
‑
2',和h3
‑
2'),一个
‑
nh
‑
co
‑
ch
‑
c
‑
基团(c
‑
2~4,h
‑
1,h
‑
3和nh)。为了满足化合物的8个不饱和度,两个羰基、一个双键、苯环和
‑
nh
‑
co
‑
ch
‑
c
‑
基团应形成喹啉
‑
2(1h)
‑
酮。该推断可进一步由h
‑
3与c
‑
2、c
‑
4、c
‑
4a,nh与c
‑
2、c
‑
3、c
‑
8、c
‑
4a、c
‑
8a,h
‑
8与c
‑
4a、c
‑
8a,h
‑
5与c
‑
4、c
‑
4a、c
‑
8a的hmbc相关得到证实。化合物的母体骨架确定后,剩余的取代基位置可通过进一步分析其hmbc相关确定。根据h
‑
2'与c
‑
4,h
‑
3与c
‑
1'的hmbc相关,可确定乙酰基取代在c
‑
4位;根据h
‑
4'与c
‑
6、c
‑
7、c
‑
8,h6
‑
5',6'与c
‑
7的hmbc相关,可确定异丙基取代在c
‑
7位。最后,根据h3
‑
3'与c
‑
5、c
‑
6、c
‑
7的hmbc相关,可确定甲基(c
‑
1')取代在c
‑
6位。至此,本发明化合物的结构得到确认,鉴定为4
‑
乙酰
‑7‑
异丙基
‑6‑
甲基喹啉
‑
2(1h)
‑
酮。
[0055]
实施例5
[0056]
取实施例2制备的化合物,为棕色胶状物。测定方法与实施例4相同,确认实施例2制备的化合物为所述的喹啉生物碱类化合物——4
‑
乙酰
‑7‑
异丙基
‑6‑
甲基喹啉
‑
2(1h)
‑
酮。
[0057]
实施例6
[0058]
取实施例3制备的化合物,为棕色胶状物。测定方法与实施例4相同,确认实施例3
制备的化合物为所述的喹啉生物碱类化合物——4
‑
乙酰
‑7‑
异丙基
‑6‑
甲基喹啉
‑
2(1h)
‑
酮。
[0059]
实施例7
[0060]
取实施例1~3制备的任一喹啉生物碱类化合物进行抗烟草花叶病毒活性试验,试验情况如下:
[0061]
采用半叶法,在药剂的质量浓度均为20m时对本发明化合物进行抗烟草花叶病毒活性测定。在5~6株烤烟的植株上,选取适用于测试的叶片(叶行正常,无病无虫),先将叶片均匀撒上细金刚砂,用毛笔将备用的烟草花叶病毒源(3.0
×
10
‑3)均匀抹在撒有金刚砂的叶片上,待所有中选的叶片接毒结束后,立即放在盛有药液的培养皿中处理20min,取出,洒去叶片上水珠和约液,将两个半叶复原排放在铺有卫生纸保湿的搪瓷衙内加盖玻璃,控温(23
±
2)℃,放在温室自然光照射,2~3d即可见枯斑.每个处理都设另一半叶为对照,另外设有1组为商品宁南霉素的处理作为对比,按下公式计算相对抑制率。
[0062]
xi%=(ck
‑
t)/ck
×
100%
[0063]
x:相对抑制率(%),ck:浸泡于清水中半片接毒叶的枯斑数(个),t浸泡于药液中半片接毒叶的枯斑数(个)。
[0064]
结果明本化合物的相对抑制率为38.5%,高于对照宁南霉素的相对抑制率33.0%,说明化合物有很好的抗烟草花叶病毒活性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1