一种瑞卢戈利及其中间体的制备方法与流程
1.本发明涉及一种瑞卢戈利及其中间体的制备方法。
背景技术:
2.2020年12月18日,美国食品药物管理局(fda)批准了瑞卢戈利orgovyx(relugolix)用于治疗晚期前列腺癌成人患者。瑞卢戈利是一种口服非肽类促性腺激素释放激素gnrh受体阻断剂,由武田、aska和myovant共同开发,2018年已经在日本获批用于子宫肌瘤治疗。除了已经获批的子宫肌瘤、前列腺癌,还在开发瑞卢戈利与诺孕酮的复方制剂,用于子宫内膜异位症引起的疼痛(phase 3)、痛经(phase 2)等治疗。
3.目前公开的瑞卢戈利的合成方法主要有以下路线。
4.(1)j.med.chem.2011,54,4998-5012,公开了如下合成路线:
[0005][0006]
该条路线最后一步收率仅44%,且总收率相对偏低。
[0007]
(2)专利cn104703992a公开了如下合成路线:
[0008]
[0009]
该条路线收率总体较高,但存在缺陷一,最后一步成脲反应,生成约2.0%的缩脲副产物,该杂质难以除去。该杂质随着投料规模放大,有增加的趋势,去除难度进一步加大。此外,该条路线中由int-5制备int-6过程中会生成难以除去的lc-ms检测含量6.1%和2.5%的杂质,含量6.1%杂质的[m+h]
+
=537.1,含量2.5%杂质的[m+h]
+
=508.1。
[0010]
(3)中国专利cn111333633a公开了如下合成路线:
[0011][0012]
该条路线收率总体较高,但存在缺陷,从int-2制备int-3过程中,氢化反应过程会产生分子量为[m+h]
+
=503.1的杂质。在后处理时难以除去。随着投料规模放大,杂质有增加的趋势,去除难度进一步加大。
技术实现要素:
[0013]
本发明所要解决的技术问题是现有的瑞卢戈利制备方法容易生成较难分离的杂质,为此,本发明提供了一种瑞卢戈利及其中间体的制备方法。本发明的制备方法可以高效地制备瑞卢戈利,并避免了难以分离杂质的生成。本发明制备方法的总收率高。
[0014]
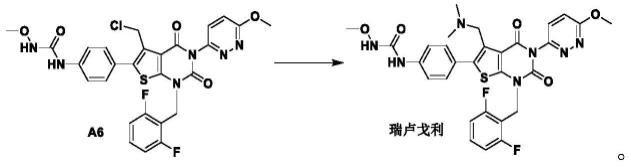
本发明提供一种瑞卢戈利的制备方法,其包括如下步骤:如式a6所示的化合物,在腈类溶剂和碱存在下,与二甲胺进行如下式所示的偶联发应得到瑞卢戈利;
[0015][0016]
所述的偶联反应中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱可为三乙胺。
[0017]
所述的偶联反应中,所述的偶联反应的反应温度为本领域此类反应的常规反应温度,较佳地,所述的偶联反应的反应温度可为25℃。
[0018]
所述的偶联反应中,所述的碱与所述的如式a6所示的化合物的摩尔比可为1.2:1。
[0019]
所述的偶联反应中,所述的腈类溶剂为本领域此类反应常规的腈类溶剂;较佳地,所述的腈类溶剂可为乙腈。
[0020]
所述的偶联反应中,所述的二甲胺可为二甲胺四氢呋喃溶液;例如2.0m二甲胺四
氢呋喃溶液。
[0021]
所述的偶联反应中,所述的二甲胺与所述的如式a6所示的化合物的摩尔比可为1.1:1。
[0022]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:乙酸乙酯中,如式a5所示的化合物与氯化试剂进行如下式所示的氯化反应得到所述的如式a6所示的化合物;
[0023][0024]
所述的氯化反应中,所述的氯化反应在引发剂存在下进行,所述的引发剂为本领域此类反应常规的引发剂。较佳地,所述的引发剂可为aibn。更佳地,所述的引发剂与所述的如式a5所示的化合物的摩尔比可为1:5。
[0025]
所述的氯化反应中,所述的氯化反应的反应温度为本领域此类反应的常规反应温度,较佳的,所述的氯化反应的反应温度可为60℃。
[0026]
所述的氯化反应中,所述的氯化试剂与所述的如式a5所示的化合物的摩尔比可为1.1:1。
[0027]
所述的氯化反应中,所述的氯化试剂为n-氯代丁二酰亚胺(ncs)。
[0028]
所述的氯化反应中,所述的如式a5所示的化合物与所述的乙酸乙酯的质量体积比可为58g/l。
[0029]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如方案1或方案2所述的步骤,
[0030]
所述的方案1包括如下步骤:溶剂中,在碱存在下,如式a4所示的化合物进行如下式所示的环化反应得到所述的如式a5所示的化合物;r为c
1-c4的烷基;
[0031][0032]
所述的方案2包括如下步骤:溶剂中,在碱存在下,如式b4所示的化合物、n,n'-羰基二咪唑(cdi)和甲氧基胺进行如下式所示的酰胺化反应得到所述的如式a5所示的化合物;
[0033]
[0034]
所述的方案1的环化反应中,所述的c
1-c4的烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基,例如正丁基。
[0035]
所述的方案1的环化反应中,所述的溶剂为本领域此类反应常规的溶剂,较佳地,所述的溶剂可为醇类溶剂和醚类溶剂的混合溶剂;所述的醇类溶剂优选为甲醇;所述的醚类溶剂优选为四氢呋喃;更佳地所述的甲醇与所述的四氢呋喃的体积比为10:1。
[0036]
所述的方案1的环化反应中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱为醇盐;所述的醇盐优选为甲醇钠;例如30%甲醇钠甲醇溶液。
[0037]
所述的方案1的环化反应中,所述的碱与所述的如式a4所示的化合物的摩尔比可为1:2。
[0038]
所述的方案1的环化反应的反应温度为本领域此类反应的常规反应温度,较佳地,所述的环化反应的反应温度可为60℃。
[0039]
所述的方案2中的酰胺化反应中,所述的溶剂为本领域此类反应常规的溶剂,较佳地,所述的溶剂为腈类溶剂;较佳地,所述的腈类溶剂可为乙腈。
[0040]
所述的方案2中的酰胺化反应中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱可为三乙胺。
[0041]
所述的方案2中的酰胺化反应的反应温度为本领域此类反应的常规反应温度,较佳地,所述的酰胺反应的反应温度可为60℃。
[0042]
所述的方案2中的酰胺反应中,所述的碱与所述的如式b4所示的化合物的摩尔比为1:1。
[0043]
所述的方案2中的酰胺化反应中,所述的n,n'-羰基二咪唑所述与如式b4所示的化合物的摩尔比为2:1。
[0044]
所述的方案2中的酰胺化反应中,所述的甲氧基胺以甲氧基胺盐形式加入反应,例如甲氧基胺盐酸盐。
[0045]
所述的方案2中的酰胺化反应中,所述的甲氧基胺与所述的如式b4所示的化合物的摩尔比为2:1。
[0046]
所述的方案2中的酰胺化反应中,所述的n,n'-羰基二咪唑和所述的甲氧基胺先混合后,再加入所述的如式b4所示的化合物。
[0047]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:溶剂中,在碱和hatu存在下,如式a3所示的化合物与3-氨基-6-甲氧基哒嗪进行如下式所示的酰胺化反应q得到所述的如式a4所示的化合物;
[0048][0049]
所述的酰胺化反应q中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱可为n,n-二异丙基乙胺。
[0050]
所述的酰胺化反应q中所述的反应温度为本领域此类反应的常规反应温度,较佳地,所述的酰胺化反应的反应温度可为80℃。
[0051]
所述的酰胺化反应q中,所述的碱与所述的如式a3所示的化合物的摩尔比可为1.5:1。
[0052]
所述的酰胺化反应q中,所述的hatu{(2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)}与所述的如式a3所示的化合物的摩尔比可为1.1:1。
[0053]
所述的酰胺化反应q中,所述的3-氨基-6-甲氧基哒嗪与所述的如式a3所示的化合物的摩尔比可为1.2:1。
[0054]
所述的酰胺化反应q中,所述的溶剂为本领域此类反应常规的溶剂,较佳地,所述的溶剂为酰胺类溶剂;更佳地,所述的酰胺类溶剂可为n,n-二甲基乙酰胺。
[0055]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:所述的如式a2所示的化合物经水解反应得到所述的如式a3所示的化合物;r-1
为c
1-c4的烷基;
[0056][0057]
所述的水解反应中,所述的c
1-c4的烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基,例如乙基。
[0058]
所述的水解反应中,所述的溶剂为乙醇。
[0059]
所述的水解反应中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱可为2.0m氢氧化钾水溶液。
[0060]
所述的水解反应中,所述的碱与所述的如式a2所示的化合物的摩尔比可为2.5:1。
[0061]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:溶剂中,在碱存在下,如式a1所示的化合物、n,n'-羰基二咪唑(cdi)和甲氧基胺进行如下式所示的酰胺化反应m得到所述的如式a2所示的化合物;
[0062][0063]
所述的酰胺化反应m中,所述的溶剂为本领域此类反应常规的溶剂,较佳地,所述的溶剂为腈类溶剂;较佳地,所述的腈类溶剂可为乙腈。
[0064]
所述的酰胺化反应m中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱可为三乙胺。
[0065]
所述的酰胺化反应m中,所述的反应温度为本领域此类反应的常规反应温度,较佳地,所述的胺化反应的反应温度可为60℃。
[0066]
所述的酰胺化反应m中,所述的碱与所述的如式a1所示的化合物的摩尔比可为1:1。
[0067]
所述的酰胺化反应m中,所述的n,n'-羰基二咪唑所述与如式a1所示的化合物的摩
尔比可为2:1。
[0068]
所述的酰胺化反应m中,所述的甲氧基胺以甲氧基胺盐形式加入反应,例如甲氧基胺盐酸盐。
[0069]
所述的酰胺化反应m中,所述的甲氧基胺与如式a1所示的化合物的摩尔比可为2:1。
[0070]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:溶剂中,在sncl
2 2h2o存在下,所述的如式sm-1所示的化合物进行如下式所示的还原反应得到如式a1所示的化合物;
[0071][0072]
所述的还原反应中,所述的溶剂可为乙醇。
[0073]
所述的还原反应中,所述的还原反应的反应温度为本领域此类反应的常规反应温度,较佳的,所述的还原反应的反应温度可为60℃。
[0074]
所述的还原反应中,所述的sncl
2 2h2o与所述的如式sm-1所示的化合物的摩尔比可为5:1。
[0075]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:溶剂中,在催化剂存在下,如式b3所示的化合物进行如下式所示的还原反应得到所述的如式b4所示的化合物;
[0076][0077]
所述的还原反应中,所述的溶剂可为甲醇和二氯甲烷的混合溶剂;较佳地,甲醇和二氯甲烷的体积比为1:3。
[0078]
所述的还原反应中,所述的催化剂为钯催化剂,例如pd/c,又例如10%pd/c(10%pd/c中含水55%)。
[0079]
所述的还原反应中,所述的还原反应的反应温度为本领域此类反应的常规反应温度,较佳地,所述的反应温度为25℃。
[0080]
所述的还原反应中,所述的催化剂与所述的如式b3所示的化合物的质量比为1:10。
[0081]
所述的还原反应中,所述的还原反应的还原剂为氢气,例如常压氢气。
[0082]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:溶剂中,在碱存在下,如式b2所示的化合物进行如下式所示的环化反应得到所述的如式b3所示的化合物;
[0083][0084]
所述的环化反应中,所述的溶剂为本领域此类反应常规的溶剂,较佳地,所述的溶剂醇类溶剂和醚类溶剂的混合溶剂;所述的醇类溶剂优选为甲醇;所述的醚类溶剂优选为四氢呋喃。
[0085]
所述的环化反应中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱为醇盐;所述的醇盐优选为甲醇钠。
[0086]
所述的环化反应中,所述的碱与所述的如式b2所示的化合物的摩尔比为1:2。
[0087]
所述的环化反应中,所述的环化反应的反应温度为本领域此类反应的常规反应温度,较佳地,所述的环化反应的反应温度为60℃。
[0088]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:溶剂中,在碱和hatu存在下,如式b1所示的化合物与3-氨基-6-甲氧基哒嗪进行如下式所示的酰胺化反应y得所述的如式b2所示的化合物;
[0089][0090]
所述的酰胺化反应y中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱可为n,n-二异丙基乙胺。
[0091]
所述的酰胺化反应y中,所述的酰胺化反应的反应温度为本领域此类反应的常规反应温度,较佳地,所述的酰胺化反应的反应温度可为80℃。
[0092]
所述的酰胺化反应y中,所述的碱与所述的如式b1所示的化合物的摩尔比可为1.5:1。
[0093]
所述的酰胺化反应y中,所述的hatu(2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)与所述的如式b1所示的化合物的摩尔比可为1.1:1。
[0094]
所述的酰胺化反应y中,所述的3-氨基-6-甲氧基哒嗪与所述的如式b1所示的化合物的摩尔比可为1.2:1。
[0095]
本所述的酰胺化反应y中,所述的溶剂为本领域此类反应常规的溶剂,较佳地,所述的溶剂为酰胺类溶剂;较佳地,所述的酰胺类溶剂为n,n-二甲基乙酰胺。
[0096]
本发明一些实施方案中,所述的瑞卢戈利的制备方法还包括如下步骤:溶剂中,在碱存在下,所述的如式sm-1所示的化合物经水解反应得到所述的如式b1所示的化合物;
[0097][0098]
所述的水解反应中,所述的溶剂可为乙醇。
[0099]
所述的水解反应中,所述的碱为本领域此类反应常规的碱;较佳地,所述的碱可为2.0m氢氧化钾水溶液。
[0100]
所述的水解反应中,所述的碱与所述的如式sm-1所示的化合物的摩尔比可为2.5:1。
[0101]
本发明还提供一种如a1所示化合物的制备方法,其包括以下步骤:溶剂中,在sncl
2 2h2o存在下,如式sm-1所示的化合物进行如下式所示的还原反应得到如式a1所示的化合物;r为c
1-c4的烷基;
[0102][0103]
较佳地,所述的如a1所示化合物的制备方法中,所述的还原反应的反应条件如前所述。
[0104]
所述的如a1所示化合物的制备方法中,所述的c
1-c4的烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基,例如正丁基。
[0105]
本发明还提供一种如a2所示化合物的制备方法,其包括以下步骤:溶剂中,在碱存在下,如式a1所示的化合物、n,n'-羰基二咪唑和甲氧基胺进行如下式所示的酰胺化反应m得到如式a2所示的化合物;r-1
为c
1-c4的烷基;r为c
1-c4的烷基;
[0106][0107]
较佳地,所述的如a2所示化合物的制备方法中,所述的酰胺化反应m的反应条件如前所述。
[0108]
所述的如a2所示化合物的制备方法中,所述的c
1-c4的烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基,例如正丁基或乙基;较佳地,r为正丁基;r-1
为乙基。
[0109]
所述的如a2所示化合物的制备方法还包括下述步骤:按照前述的如a1所示化合物的制备方法制备得到所述的如式a1所示的化合物。
[0110]
本发明还提供一种如a3所示化合物的制备方法,其包括以下步骤:所述的如式a2
所示的化合物经水解反应得到如式a3所示的化合物;r-1
为c
1-c4的烷基;r为c
1-c4的烷基;
[0111][0112]
较佳地,所述的如a3所示化合物的制备方法中,所述的水解反应的反应条件如前所述。
[0113]
所述的如a3所示化合物的制备方法中,所述的c
1-c4的烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基,例如正丁基或乙基;较佳地,r为正丁基;r-1
为乙基。
[0114]
所述的如a3所示化合物的制备方法还包括下述步骤:按照前述的如式a2所示的化合物的制备方法制备得到所述的如a2所示化合物。
[0115]
本发明还提供一种如a4所示化合物的制备方法,溶剂中,在碱和hatu存在下,如式a3所示的化合物与3-氨基-6-甲氧基哒嗪进行如下式所示的酰胺化反应q得到如式a4所示的化合物;r为c
1-c4的烷基;
[0116][0117]
较佳地,所述的如a4所示化合物的制备方法中,所述的酰胺化反应q的条件如前所述。
[0118]
所述的如a4所示化合物的制备方法中,所述的c
1-c4的烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基,例如正丁基。
[0119]
所述的如a4所示化合物的制备方法还包括下述步骤:按照前述的如式a3所示的化合物的制备方法制备得到所述的如a3所示化合物。
[0120]
本发明还提供一种如a5所示化合物的制备方法,其为方案1或方案2;
[0121]
所述的方案1包括如下步骤:溶剂中,在碱存在下,如式a4所示的化合物进行如下式所示的环化反应得到如式a5所示的化合物;r为c
1-c4的烷基;
[0122][0123]
所述的方案2包括如下步骤:溶剂中,在碱存在下,如式b4所示的化合物、n,n'-羰基二咪唑(cdi)和甲氧基胺进行如下式所示的酰胺化反应得到如式a5所示的化合物;
[0124][0125]
所述的如a5所示化合物的制备方法中,所述的c
1-c4的烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基,例如正丁基。
[0126]
所述的如a5所示化合物的制备方法中,所述的方案1中的环化反应的反应条件如前所述。
[0127]
所述的如a5所示化合物的制备方法中,所述的方案2中的酰胺化反应的反应条件如前所述。
[0128]
所述的如a5所示化合物的制备方法还包括下述步骤:按照前述所述的如式a4所示的化合物的制备方法制备得到所述的如a4所示化合物。
[0129]
所述的如a5所示化合物的制备方法还包括下述步骤:按照前本发明所述的如式b4所示的化合物的制备方法制备得到所述的如b4所示化合物。
[0130]
本发明还提供一种如b1所示化合物的制备方法,其包括以下步骤:溶剂中,在碱存在下,如式sm-1所示的化合物经水解反应得到如式b1所示的化合物;r为正丁基;r-1
为乙基;
[0131][0132]
较佳地,所述的如b1所示化合物的制备方法中,所述的水解反应的反应条件如前所述。
[0133]
本发明还提供一种如b2所示化合物的制备方法,其包括以下步骤:溶剂中,在碱和hatu存在下,如式b1所示的化合物与3-氨基-6-甲氧基哒嗪进行如下式所示的酰胺化反应y得到如式b2所示的化合物;
[0134][0135]
较佳地,所述的如b2所示化合物的制备方法中,所述的酰胺化反应y的反应条件如前所述。
[0136]
所述的如b2所示化合物的制备方法还包括下述步骤:按照前述所述的如式b1所示的化合物的制备方法制备得到所述的如b1所示化合物。
[0137]
本发明还提供一种如b3所示化合物的制备方法,其包括以下步骤:溶剂中,在碱存
在下,如式b2所示的化合物进行如下式所示的环化反应得到如式b3所示的化合物;
[0138][0139]
较佳地,所述的如b3所示化合物的制备方法中,所述的环化反应的反应条件如前所述。
[0140]
所述的如b3所示化合物的制备方法还包括下述步骤:按照前述所述的如式b2所示的化合物的制备方法制备得到所述的如b2所示化合物。
[0141]
本发明还提供一种如b4所示化合物的制备方法,其包括以下步骤:溶剂中,在催化剂存在下,如式b3所示的化合物进行如下式所示的还原反应得到如式b4所示的化合物;
[0142][0143]
较佳地,所述的如b4所示化合物的制备方法中,所述的还原反应的反应条件如前所述。
[0144]
所述的如b4所示化合物的制备方法还包括下述步骤:按照前述所述的如式b3所示的化合物的制备方法制备得到所述的如b3所示化合物。
[0145]
本发明还提供一种如a1、a2、a3、a4、a5、b1、b2或b4所示化合物:
其中r和r-1
如前所述;
[0146]
较佳地,a4为a1为a2为或a3为
[0147]
本发明还提供一种如前所述的如a1、a2、a3、a4、a5、b1、b2或b4所示化合物在制备瑞卢戈利中的应用。
[0148]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0149]
本发明所用试剂和原料均市售可得。
5。分子量是[m+h]
+
=503.1,解析结构如下式所示由于结构与其反应中间体3的结构极为相似,在后处理时难以除去。随着投料规模放大,杂质有增加的趋势,去除难度进一步加大。发明人在分析和探索产生以上杂质的原因时发现,氢化步骤容易产生较多的杂质且不易除去。因此,为了避免上述杂质的产生,本发明通过改变骨架片段的合成顺序,开发了一条总体收率高的瑞卢戈利工艺路线。
[0155]
本技术实施例以r为正丁基,r-1
为乙基为例,说明制备瑞卢戈利的方法,以及中间体a1、a2、a3、a4、a5、b1、b2、b3和b4的制备方法。
[0156]
实施例1制备中间体a1
[0157][0158]
室温条件下,将sm-1(106.4g,0.20mol,1.0eq.)和sncl
2 2h2o(225.6g,1.0mol,5.0eq.)依次加入到乙醇(1.2l)中,反应液升温至60℃后搅拌2小时。lc-ms监测反应结束后,将反应液的溶剂乙醇减压除去后,依次放入二氯甲烷(2.0l)和1.0m氢氧化钠溶液(2.0l),搅拌15分钟,过滤,滤饼用二氯甲烷(1.0l)洗涤,收集有机相,水相继续用二氯甲烷(2.0l)萃取,收集并合并有机相,通过无水硫酸钠干燥,过滤,旋干得到中间体a1(淡黄色油状物;98.5g;产率:98%)。1h-nmr(600mhz,dmso-d6,δppm):7.43-7.38(m,1h),7.06(t,j=7.8hz,2h),6.98(d,j=8.4hz,2h),6.59(d,j=8.4hz,2h),5.37(s,2h),4.86(s,2h),4.14(q,j=6.6hz,2h),4.03(t,j=7.2hz,2h),2.23(s,3h),1.50-1.40(m,2h),1.23-1.16(m,5h),0.88-0.79(m,3h).esi(m/z):[m+h]
+
:503.1.
[0159]
实施例2制备中间体a2
[0160][0161]
将cdi(58.4g,0.36mol,2.0eq.)和三乙胺(18.2g,0.18mol,1.0eq.)依次加入到乙腈(0.5l)中,在25℃条件下反应液搅拌15分钟,向其中分批加入甲氧基胺盐酸盐(30.0g,
0.36mol,2.0eq.),加料完成后在此温度下搅拌1小时,再加入中间体a1(90.4g,0.18mol,1.0eq.),反应液升温至60℃后搅拌2小时。lc-ms监测反应结束后,将反应液的溶剂乙腈减压除去后,加入乙酸乙酯(1.2l)和水(1.2l),萃取,分层,收集有机相,并浓缩至大约0.3l时,加入正庚烷(0.6l),将该混合物在10℃下搅拌2小时,过滤并干燥后得到中间体a2(淡黄色固体;101.6g;产率:98%)。1h-nmr(600mhz,dmso-d6,δppm):9.60(s,1h),9.05(s,1h),7.68(d,j=8.4hz,2h),7.42-7.40(m,1h),7.25(d,j=6.6hz,2h),7.06(t,j=7.8hz,2h),4.88(s,2h),4.15(q,j=6.6hz,2h),4.03(t,j=7.2hz,2h),3.63(s,3h),2.27(s,3h),1.55-1.43(m,2h),1.23-1.16(m,5h),0.88-0.79(m,3h).esi(m/z):[m+h]
+
:576.1.
[0162]
实施例3制备中间体a3
[0163][0164]
将中间体a2(97.8g,0.17mol,1.0eq.)和2.0m氢氧化钾水溶液(0.21l,0.425mol,2.5eq.)依次加入到乙醇(0.8l)中,反应液升温至60℃后搅拌8小时。lc-ms监测反应结束后,将反应液的溶剂乙醇减压除去后,加入水(0.5l)和12n盐酸(80ml),调节反应液ph值为2左右。加入乙酸乙酯(1.0l),萃取,分层,收集有机相,并浓缩至大约0.3l时,加入正庚烷(0.7l),将该混合物在10℃下搅拌2小时,过滤并干燥后得到中间体a3(淡黄色固体;89.3g;产率:96%)。1h-nmr(600mhz,dmso-d6,δppm):12.93(s,1h),9.59(s,1h),9.04(s,1h),7.66(d,j=9.0hz,2h),7.42-7.38(m,1h),7.22(d,j=7.8hz,2h),7.06(t,j=7.8hz,2h),4.88(s,2h),4.03(t,j=7.2hz,2h),3.62(s,3h),2.29(s,3h),1.55-1.44(m,2h),1.23-1.16(m,2h),0.86-0.80(m,3h).esi(m/z):[m+h]
+
:548.1.
[0165]
实施例4制备中间体a4
[0166][0167]
将中间体a3(87.6g,0.16mol,1.0eq.),3-氨基-6-甲氧基哒嗪(24.0g,0.192mol,1.2eq.),n,n-二异丙基乙胺(31.0g,0.24mol,1.5eq.)和hatu(66.8g,0.176mol,1.1eq.)依次加入到n,n-二甲基乙酰胺(0.8l)中,反应液升温至80℃后搅拌5小时。lc-ms监测反应结束后,将反应液自然降温至室温后,加入水(0.8l)和乙酸乙酯(0.8l),萃取,分层,收集有机相,并浓缩至大约0.3l时,加入正庚烷(0.6l),将该混合物在10℃下搅拌2小时,过滤并干燥后得到中间体a4(白色固体;99.5g;产率:95%)。1h-nmr(600mhz,dmso-d6,δppm):10.60(s,1h),9.60(s,1h),9.06(s,1h),8.20(d,j=9.0hz,1h),7.70(d,j=9.0hz,2h),7.42-7.37(m,1h),7.31-7.29(m,3h),7.04(t,j=8.4hz,2h),4.97(s,2h),4.06(t,j=6.6hz,2h),4.00(s,3h),3.63(s,3h),2.24(s,3h),1.50-1.45(m,2h),1.18(q,j=7.2hz,2h),0.75(t,j
=7.2hz,3h).esi(m/z):[m+h]
+
:655.2.
[0168]
实施例5制备中间体a5
[0169][0170]
将中间体a4(98.1g,0.15mol,1.0eq.)和30%甲醇钠甲醇溶液(13.5g,0.075mol,0.5eq.)依次加入到四氢呋喃(0.15l)和甲醇(1.5l)混合溶液中,反应液升温至60℃后搅拌1小时。lc-ms监测反应结束后,将反应液自然降温至室温后,向其中加入12n盐酸(6.25ml),调节反应液ph值为中性,将该混合物减压浓缩至大约1.0l后,在10℃下搅拌2小时,过滤并干燥后得到中间体a5(白色固体;85.3g;产率:98%)。1h-nmr(600mhz,dmso-d6,δppm):9.70(s,1h),9.25(s,1h),7.75(d,j=9.0hz,1h),7.70(d,j=8.4hz,2h),7.46(d,j=9.0hz,2h),7.35(d,j=8.4hz,2h),7.13(t,j=8.4hz,2h),5.38(d,j=13.8hz,1h),5.17(d,j=13.8hz,1h),4.09(s,3h),3.63(s,3h),2.40(s,3h).esi(m/z):[m+h]
+
:581.1.
[0171]
实施例6制备中间体a6
[0172][0173]
将中间体a5(87.0g,0.15mol,1.0eq.),n-氯代丁二酰亚胺(22.0g,0.165mol,1.1eq.)和aibn(4.92g,0.03mol,0.2eq.)依次加入到乙酸乙酯(1.5l)中,反应液升温至60℃后搅拌30分钟。lc-ms监测反应结束后,将反应液自然降温至温室后,向其中加入水(1.5l),萃取,分层,收集有机相,并浓缩至大约0.5l时,将该混合物在10℃下搅拌2小时,过滤并干燥后得到中间体a6(白色固体;85.0g;产率:92%)。1h-nmr(400mhz,dmso-d6,δppm):9.70(s,1h),9.16(s,1h),7.81-7.76(m,3h),7.49-7.46(m,4h),7.15(t,j=8.0hz,2h),5.36(s,1h),5.23(s,1h),4.86(s,2h),4.10(s,3h),3.64(s,3h).esi(m/z):[m+h]
+
:615.1&617.1.
[0174]
实施例7制备中间体b1
[0175][0176]
将中间体sm-1(106.4g,0.20mol,1.0eq.)和2.0m氢氧化钾水溶液(0.25l,0.50mol,2.5eq.)加入到乙醇(0.8l)中,反应液升温至60℃后搅拌2小时。lc-ms监测反应结束后,将反应液的溶剂乙醇减压除去后,加入水(0.3l)和12n盐酸(60ml),调节反应液的ph
值至2左右,加入乙酸乙酯(0.8l),萃取,分层,收集有机相,并浓缩至大约0.3l时,加入正庚烷(0.6l),将该混合物在10℃下搅拌2小时,过滤并干燥后得到中间体b1(类白色固体;98.8g;产率:98%)。1h-nmr(400mhz,dmso-d6,δppm):13.05(s,1h),8.27(d,j=8.8hz,2h),7.63(d,j=8.0hz,2h),7.45-7.37(m,1h),7.07(t,j=8.0hz,2h),4.90(s,2h),4.03(t,j=7.2hz,2h),2.36(s,3h),1.55-1.45(m,2h),1.23-1.15(m,2h),0.86-0.80(m,3h).esi(m/z):[m+h]
+
:505.1.
[0177]
实施例8制备中间体b2
[0178][0179]
将中间体b1(95.8g,0.19mol,1.0eq.),3-氨基-6-甲氧基哒嗪(28.5g,0.228mol,1.2eq.),n,n-二异丙基乙胺(36.8g,0.285mol,1.5eq.)和hatu(79.4g,0.209mol,1.1eq.)依次加入到n,n-二甲基乙酰胺(1.0l)中,反应液升温至80℃后搅拌5小时。lc-ms监测反应结束后,将反应液自然降温至室温后,加入水(1.0l)和乙酸乙酯(1.0l),萃取,分层,收集有机相,并浓缩至大约0.35l时,加入正庚烷(0.7l),将该混合物在10℃下搅拌2小时,过滤并干燥后得到中间体b2(白色固体;108.0g;产率:93%)。1h-nmr(400mhz,dmso-d6,δppm):10.79(s,1h),8.31(d,j=8.8hz,2h),8.21(d,j=9.6hz,1h),7.69(d,j=8.8hz,2h),7.45-7.37(m,1h),7.31(d,j=9.6hz,1h),7.06(t,j=8.0hz,2h),4.99(s,2h),4.05(t,j=6.8hz,2h),4.00(s,3h),2.32(s,3h),1.50-1.43(m,2h),1.17(q,j=7.6hz,2h),0.74(t,j=7.6hz,3h).esi(m/z):[m+h]
+
:612.1.
[0180]
实施例9制备中间体b3
[0181][0182]
将中间体b2(103.9g,0.17mol,1.0eq.)和30%甲醇钠甲醇溶液(15.3g,0.085mol,0.5eq.)依次加入到四氢呋喃(0.15l)和甲醇(1.5l)混合溶液中,反应液升温至60℃后搅拌1小时。lc-ms监测反应结束后,将反应液自然降温至室温后,向其中加入12n盐酸(7.0ml),调节反应液ph值为中性,将该混合物减压浓缩至大约1.0l后,在10℃下搅拌2小时,过滤并干燥后得到中间体b3(淡黄色固体;89.5g;产率:98%)。1h-nmr(600mhz,dmso-d6,δppm):8.32(d,j=9.0hz,2h),7.77-7.74(m,3h),7.47(d,j=9.0hz,2h),7.14(t,j=8.4hz,2h),5.39(s,1h),5.22(s,1h),4.09(s,3h),2.50(s,3h).esi(m/z):[m+h]
+
:538.0.
[0183]
实施例10制备中间体b4
[0184][0185]
将中间体b3(85.9g,0.16mol,1.0eq.)和10%pd/c(8.59g,含水55%)加入到二氯甲烷(1.2l)和甲醇(0.4l)的混合溶液中,将反应体系置换成h2氛围后,在25℃下搅拌24个小时。lc-ms监测反应结束后,滤除催化剂,并用二氯甲烷(0.3l)洗涤,将滤液压浓缩至大约0.35l后加入甲醇(0.3l),在10℃下搅拌2小时,过滤并干燥后得到中间体b4(白色固体;78.0g;产率:96%)。1h-nmr(600mhz,dmso-d6,δppm):7.75(d,j=9.0hz,1h),7.46(d,j=9.0hz,2h),7.14(t,j=8.4hz,2h),7.09(d,j=8.4hz,2h),6.64(d,j=8.4hz,2h),5.43(s,2h),5.38(d,j=15.0hz,1h),5.17(d,j=15.0hz,1h),4.10(s,3h),2.36(s,3h).esi(m/z):[m+h]
+
:508.1.
[0186]
实施例11制备中间体a5
[0187][0188]
将cdi(48.6g,0.30mol,2.0eq.)和三乙胺(15.2g,0.15mol,1.0eq.)依次加入到乙腈(0.5l)中,25℃条件下反应液搅拌15分钟,向其中分批加入甲氧基胺盐酸盐(25.0g,0.30mol,2.0eq.),加料完成后,在此温度下搅拌1小时后,再加入中间体b4(76.0g,0.15mol,1.0eq.),将反应液升温至60℃后搅拌2小时。lc-ms监测反应结束后,将反应液压浓缩至大约0.15l后加入甲醇(0.8l),在10℃下搅拌2小时,过滤并干燥后得到中间体a5(白色固体;82.8g;产率:95%)。1h-nmr(600mhz,dmso-d6,δppm):9.70(s,1h),9.25(s,1h),7.75(d,j=9.0hz,1h),7.70(d,j=8.4hz,2h),7.46(d,j=9.0hz,2h),7.35(d,j=8.4hz,2h),7.13(t,j=8.4hz,2h),5.38(d,j=13.8hz,1h),5.17(d,j=13.8hz,1h),4.09(s,3h),3.63(s,3h),2.40(s,3h).esi(m/z):[m+h]
+
:581.1.
[0189]
实施例12制备瑞卢戈利
[0190][0191]
将实施6制备得到的中间体a6(80.0g,0.13mol,1.0eq.),2.0m二甲胺四氢呋喃溶液(0.071l,0.143mol,1.1eq.)和三乙胺(15.7g,0.156mol,1.2eq.)依次加入到乙腈(0.8l)中,反应液在25℃搅拌2小时。lc-ms监测反应结束后,将该混合物减压浓缩后,加入水(0.8l)和二氯甲烷(0.8l),萃取,分层,收集有机相,并浓缩至大约0.35l时,加入乙酸乙酯
(0.3l),在室温条件下打浆24小时,过滤并干燥后得到瑞卢戈利(白色固体;79.3g;纯度:99.82%;产率:98%)。1h-nmr(400mhz,dmso-d6,δppm):9.63(s,1h),9.07(s,1h),7.76-7.71(m,3h),7.53-7.44(m,4h),7.14(t,j=8.0hz,2h),5.38(s,1h),5.22(s,1h),4.09(s,3h),3.64-3.53(m,5h),2.04(s,6h).esi(m/z):[m+h]
+
:624.1
[0192]
本专利得到的瑞卢戈利的化学纯度:99.82%,未检测到上述的相关杂质rs-2和rs-4。hplc条件如表1所示:
[0193]
表1
[0194][0195]
对比例1
[0196]
按照中国专利cn104703992a制备瑞卢戈利。
[0197]
将乙腈(30ml)和1,1-羰基二咪唑(cdi,5.01g,30.9mmol,1.7eq)加入到反应器中,并搅拌该混合物。在搅拌下,向其中加入三乙胺(1.56g,15.4mmol,0.85eq),并冷却至内部温度10
±
5℃。在搅拌下,在30℃或更低的内部温度下,向其中逐份加入盐酸甲氧基胺(2.90g,34.7mmol,1.91eq),并将试剂所使用的容器用乙腈(5ml)洗涤。在25
±
5℃的内部温度下搅拌该混合物,确定该混合物溶解之后,将该溶液再搅拌10分钟或更长时间。然后,在搅拌下,向其中加入6-(4-氨基苯基)-1-(2,6-二氟苄基)-5-二甲基氨基甲基-3-(6-甲氧基哒嗪-3-基)噻吩并[2,3-d]嘧啶-2,4(1h,3h)-二酮(10.0g,18.1mmol),并将试剂所使用的容器用乙腈(5ml)洗涤。将该反应混合物升温至内部温度50
±
5℃,并在相同温度下搅拌2小时,得到反应混合物称为反应混合物的。在搅拌下,在50
±
5℃的内部温度下,向其中加入三
乙胺(2.35g,23.2mmol,1.28eq)。在40-55℃的内部温度下,向其中逐滴加入自来水(40ml),将该混合物搅拌1小时,并在40-55℃的内部温度下,向其中再次逐滴加入自来水(100ml)。在搅拌下,将该混合物在25
±
5℃的内部温度下老化1小时或更长时间。过滤收集晶体,用自来水(16ml)和乙腈(4ml)的混合溶剂洗涤,得到潮湿的晶体,干燥得瑞卢戈利。其的hplc谱图如图1所示,检测条件如上表1所示。瑞卢戈利的出峰时间:12.13min(含量为99.22%),杂质rs-4的出峰时间:18.15min(含量为0.78%)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1