一种靶向PD-L1基因的小分子抑制剂及其应用的制作方法
本发明涉及一种靶向pd-l1基因的小分子抑制剂及其应用。
背景技术:
在20世纪,当人们研究纺锤霉素和偏端霉素时,他们发现一类小分子化合物可以特异性地识别dna分子并在双链dna分子的小沟处结合。受这两种小分子化合物的启发,加州理工学院的perterdervan研究团队首先设计并合成了由氨基酸、n-甲基吡咯(py)和n-甲基咪唑(im)五元芳环通过酰胺键连接而成的分子,通称:吡咯-咪唑聚酰胺(py-impolyamide)。它们可以特异性识别dna碱基序列并结合,具有良好的细胞膜通透性。perterdervan研究小组揭示了吡咯-咪唑聚酰胺识别dna序列的具体规则,即im/py和py/im对识别g·c和c·g碱基,而py/py对识别t·a或a·t碱基。随后本实验室张文等报道了(s)-β-羟基-γ-氨基丁酸/β-丙氨酸配对可以区别t·a或a·t碱基。最新的研究发现,fukuda团队首次报道了吡咯-咪唑聚酰胺可以靶向tgf-β1启动子并有效缓解进行性肾病。此外,fukuda和sugiyama的研究团队与一家日本制药公司合作进行临床研究:聚酰胺靶向tgf-β1基因治疗伤口瘢痕修复,并预测聚酰胺将用于临床治疗的基因靶向药物。
肿瘤免疫治疗是指通过调节机体免疫系统来抑制和杀死肿瘤细胞,2013年被science杂志列为年度“十大科学突破榜首”。肿瘤通过调节t淋巴细胞功能的抑制途径来逃避抗肿瘤免疫,称为免疫检查点。程序性死亡受体-1(pd-1)及其配体pd-l1是免疫治疗中最有希望的免疫检查点。pd-1(也称为cd279)是i型跨膜受体在t细胞,b细胞,单核细胞,自然杀伤t细胞和树突细胞的细胞表面表达。pd-l1经常在不同类型肿瘤的表面上过表达,包括淋巴瘤,黑色素瘤,肺癌,卵巢癌,肾肿瘤和膀胱癌等。在肿瘤细胞中,pd-1与pd-l1结合会抑制t淋巴细胞的增殖,细胞因子的释放,其导致肿瘤特异性t细胞的衰竭和凋亡,从而为癌细胞提供避免免疫应答的机会。所以阻断pd-1/pd-l1相互作用可以恢复t细胞的活力,并且可以有效杀死癌细胞。这种方法的实用性已在临床实验中得到证实,并且近年来取得了巨大的成功。美国食品和药物管理局(fda)已经批准了两种抗pd-1抗体:nivolumab(bms)用于晚期鳞状非小细胞肺癌和pembrolizumab(merck)用于晚期非小细胞肺癌和黑色素瘤等,以及三种抗pd-l1抗体,atezolizumab(genentech/roche)用于晚期膀胱癌,durvalumab(astrazeneca)用于晚期膀胱癌和avelumab(emdserono,inc)用于皮肤癌以及晚期膀胱癌;中国企业也先后开发上市5种pd-(l)1抗体,这些抗体的应用为患有癌症的患者提供了治愈的希望。但这些抗体也存在一些目前还无法解决的问题,比如:对患者差异性应答明显、总体应答率不足30%,抗体的免疫原性,只能注射难于口服,保存、运输困难及成本较高等。
基于肿瘤在全世界范围的发病率和死亡率以及抗体免疫疗法面临问题,发明小分子免疫抗肿瘤药物势在必行。由于小分子抑制剂与抗体相比,具有以下几个优点,即:降低生产成本,提高稳定性,改善肿瘤渗透性,口服给药的顺利性和消除免疫原性问题。因此开发直接靶向pd-1/pd-l1的小分子抑制剂更具有潜力和收益。然而,pd-l1小分子抑制剂的研发远远滞后于抗体,还没有一个上市的药物。主要是因为之前pd-1/pd-l1复合物的晶体结构信息不完整,以及pd-1/pd-l1相互作用表面高度疏水且相对平整。目前bristol-myerssquibb(bms)专利报道他们设计的小分子化合物被证明可以阻断pd-1/pd-l1的相互作用。这类化合物在体外作用于pd-l1,并诱导形成二聚体,阻止了与pd-1结合形成复合物来发挥作用,避免免疫逃逸。其他对于这个靶点研究的小分子化合物近来也有少量报道,但还没有应用于临床中,所以,发明小分子pd-l1基因的抑制剂充满希望又具有巨大挑战性。
通过以上对本发明相关的背景资料的检索分析发现:聚酰胺具有被开发为一种靶向pd-l1基因的小分子抑制剂的巨大潜力。
技术实现要素:
针对现有技术存在的上述技术问题,本发明的目的在于提供一种靶向pd-l1基因的小分子抑制剂及其应用。
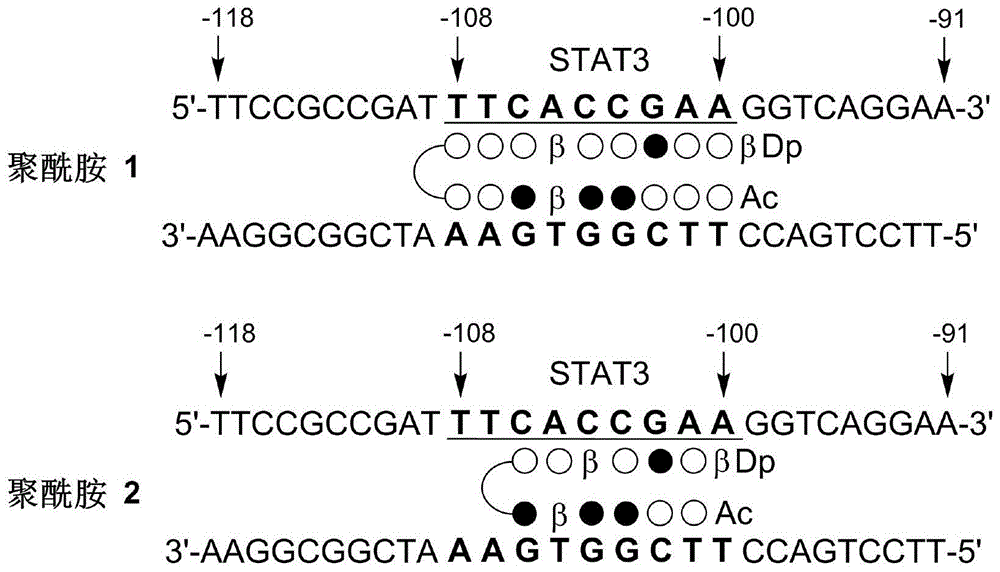
所述的一种靶向pd-l1基因的小分子抑制剂,其特征在于所述靶向pd-l1基因的小分子抑制剂为吡咯-咪唑聚酰胺类化合物,其结构式如聚酰胺1、聚酰胺2中的任意一种所示,本申请中采用错配聚酰胺为阴性对照:
其中,●:n-甲基咪唑残基;○:n-甲基吡咯残基;
β表示β-丙氨酸残基;(表示γ-氨基酸残基;
ac表示乙酰基;dp表示n,n-二甲氨基丙二胺残基。
所述的靶向pd-l1基因的小分子抑制剂在制备抗肿瘤药物中的应用。
所述的靶向pd-l1基因的小分子抑制剂在制备抗肿瘤药物中的应用,其特征在于所述小分子抑制剂用于制备以pd-l1基因为靶标的抗肿瘤药物。
进一步地,所述聚酰胺1、聚酰胺2分别与靶向pd-l1基因的启动子区dna序列结合模式如下图1中所示。结合图1中可以看出,聚酰胺1、聚酰胺2等小分子抑制剂可以通过如图1中所示的结合模式来制备治疗非小细胞肺癌或乳腺癌的抗肿瘤药物。另外,本发明的小分子抑制剂,通过靶向调节pd-l1基因表达、通过免疫和非免疫途径抗肿瘤的过程示意图如图2所示。
相对于现有技术,本发明取得的有益效果是:
本发明自主设计并合成了吡咯-咪唑聚酰胺类化合物作为小分子抑制剂,首次通过识别配对的方式竞争性结合pd-l1基因的启动子区域中stat3蛋白的结合序列,下调pd-l1基因的转录和翻译水平。从而,促进细胞因子白介素-2的分泌,增强t细胞介导的杀伤作用,在一定程度上抑制肿瘤免疫逃逸,这是通过免疫途径起到抗肿瘤的作用。另一方面,本发明设计的吡咯-咪唑聚酰胺类化合物对肿瘤细胞单独产生直接的毒性作用,抑制akt/caspase-3蛋白的表达,抑制细胞迁移,最终引起肿瘤细胞凋亡,这是通过非免疫途径聚酰胺起到抗肿瘤作用。通过实验,本发明设计的吡咯-咪唑聚酰胺类化合物具有较好的抗肿瘤性能。
附图说明
图1为本发明的聚酰胺与靶标pd-l1基因启动子区dna序列作用模式示意图;
图2为本发明的聚酰胺通过靶向调节pd-l1基因表达、通过免疫和非免疫途径抗肿瘤的过程示意图;
图3为制备聚酰胺1和2、错配聚酰胺所使用的单体或二聚体;
图4为本发明的聚酰胺1和2与pd-l1启动子区域匹配的双链-dna序列选择性相互作用图;
图5为本发明的聚酰胺与stat3蛋白竞争性结合实验结果图;
图6为本发明的聚酰胺与pd-l1双链dna的分子模拟结合示意图;
图7中a、b分别为本发明的聚酰胺对hcc827细胞和mda-mb-231细胞中pd-l1mrna表达水平的抑制图;
图8中a、b分别为本发明的聚酰胺对hcc827细胞和mda-mb-231细胞中pd-l1蛋白表达水平的抑制图;
图9中分图a为本发明的聚酰胺抑制hcc827细胞增殖和(或)促进凋亡实验数据图;
图9中分图b为本发明的聚酰胺抑制mda-mb-231细胞增殖和(或)促进凋亡实验数据图;
图9中分图c为本发明的聚酰胺抑制mrc-5细胞增殖和(或)促进凋亡实验数据图;
图10为本发明的聚酰胺对hcc827细胞和mda-mb-231细胞迁移的抑制率图;
图11中a、b分别为本发明的聚酰胺在jurkatt细胞存在下对两种肿瘤细胞和共培养体系中il-2表达量影响结果图;
图11中c、d分别为本发明的聚酰胺对两种肿瘤细胞和jurkat细胞共培养体系中介导的杀伤作用结果图。
具体实施方式
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
实施例1:聚酰胺1的制备
所使用的初始原料和试剂试剂的缩写如表1。
表1:固相合成反应中所用试剂及其缩写
合成步骤如下:
1)活化:称取图3如式(9)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3中如式(1)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3中如式(6)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,每次进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3中如式(5)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2),对步骤10)的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3中如式(5)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)重复步骤4)、步骤2)的操作,对步骤12)中的相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
14)偶合:称取图3中如式(1)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
15)重复步骤4)、步骤2)的操作,对步骤14)中的相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
16)偶合:称取图3中如式(4)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
17)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤16)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
18)偶合:称取图3中如式(5)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
19)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤18)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
20)偶合:称取图3中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
21)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤20)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
22)偶合:称取图3中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
23)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤22)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
24)偶合:称取图3中如式(8)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
25)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤24)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
26)偶合:称取图3中如式(1)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
27)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤26)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
28)偶合:称取图3中如式(5)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
29)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤28)的反应液进行同样的盖帽、脱保护、盖帽反应,获得聚酰胺1的中间体(聚酰胺1a)。为了验证合成产物,进行与步骤4)相应过程相同。
30)将步骤29)的中间体,连有目标聚酰胺1的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤、纯化后,得到淡黄色粉末的目标产物,聚酰胺1,收率:8.1%。分析hplc:tr=9.98min、98%纯度.1h-nmr(400mhz,dmso-d6):δ10.50(s,1h,芳香的nh),10.46(s,2h,芳香的nh),10.42(s,1h,芳香的nh),10.33(s,2h,芳香的nh),10.26(s,1h,芳香的nh),10.08(s,2h,芳香的nh),9.86-9.78(m,3h,芳香的nh),9.73(s,2h,芳香的nh),9.62(s,2h,芳香的nh),8.01-7.87(m,3h,脂肪的nh),7.82-7.75(m,2h,脂肪的nh),7.46(s,1h,芳香的ch),7.43-7.31(m,4h,芳香的ch),7.27-7.23(m,2h,芳香的ch),7.20(s,1h,芳香的ch),7.14-6.98(m,4h,芳香的ch),6.96-6.89(m,6h,芳香的ch),6.83-6.76(m,2h,芳香的ch),6.73-6.67(m,4h,芳香的ch),6.62-6.57(m,4h,芳香的ch),3.98(s,3h,nch3),3.95(s,3h,nch3),3.92(s,3h,2×nch3),3.88(s,3h,nch3),3.85(s,6h,2×nch3),3.81(s,3h,nch3),3.77(s,6h,2×nch3),3.74(s,6h,2×nch3),3.70(s,6h,2×nch3),3.65(s,6h,2×nch3),3.57-3.50(m,4h,2×nhch2),3.44(m,2h,nhch2),3.36-3.28(m,4h,2×nhch2),2.65(m,2h,nme2ch2),2.34(t,3jh,h=7.2hz,2h,coch2),2.28(t,3jh,h=6.8hz,2h,coch2),2.22-2.15(m,4h,2×coch2),2.04(s,6h,n(ch3)2),1.97(s,1h,coch3),1.77(m,2h,ch2ch2ch2),1.52(m,2h,ch2ch2ch2).tof-mass:m/zcalcdforc112h131n42o21[m+h]+:2400.0474;found:2400.8130[m+h]+,2422.8110[m+na]+.
实施例2:聚酰胺2的制备
合成步骤类似合成聚酰胺1,具体步骤如下:
1)活化:称取图3如式(9)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3中如式(1)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3中如式(7)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,每次进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3中如式(5)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2),对步骤10)的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3中如式(4)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)重复步骤4)、步骤2)的操作,对步骤12)中的相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
14)偶合:称取图3中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
15)重复步骤4)、步骤2)的操作,对步骤14)中的相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
16)偶合:称取图3中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
17)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤16)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
18)偶合:称取图3中如式(7)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
19)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤18)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
20)偶合:称取图3中如式(5)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
21)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤20)的反应液进行同样的盖帽、脱保护、盖帽反应,得聚酰胺2的中间体(聚酰胺2a)。为了验证合成产物,进行与步骤4)相应过程相同。
22)将步骤21)的中间体,连有目标聚酰胺2的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤、纯化后,得到灰白色粉末的目标产物,聚酰胺2,收率:13.4%。分析hplc:tr=10.94min、98%纯度.1h-nmr(400mhz,dmso-d6):δ10.43(s,1h,芳香的nh),10.37(s,1h,芳香的nh),10.30(s,2h,芳香的nh),10.13(s,1h,芳香的nh),9.97(s,2h,芳香的nh),9.85(s,2h,芳香的nh),9.71(s,1h,芳香的nh),8.26-8.00(m,3h,脂肪的nh),7.85-7.76(m,2h,脂肪的nh),7.61(s,1h,芳香的ch),7.50(s,1h,芳香的ch),7.42(s,1h,芳香的ch),7.30(s,1h,芳香的ch),7.21-6.97(m,6h,芳香的ch),6.89-6.82(m,2h,芳香的ch),6.76-6.63(m,4h,芳香的ch),3.99(s,3h,nch3),3.94(s,3h,nch3),3.90(s,6h,2×nch3),3.85(s,3h,nch3),3.80(s,6h,2×nch3),3.76(s,3h,nch3),3.72(s,6h,2×nch3),3.60(m,2h,nhch2),3.54(m,2h,nhch2),3.48(m,2h,nhch2),3.39-3.27(m,4h,2×nhch2),2.61(m,2h,nme2ch2),2.42(d,3jh,h=7.4hz,2h,coch2),2.31(t,3jh,h=6.8hz,2h,coch2),2.20-2.10(m,4h,2×coch2),2.02(s,6h,n(ch3)2),1.97(s,1h,coch3),1.77(m,2h,ch2ch2ch2),1.53(m,2h,ch2ch2ch2).tof-mass:m/zcalcdforc76h95n30o15[m+h]+:1667.7593;found:1667.5201[m+h]+,1689.4670[m+na]+.
实施例3:错配聚酰胺的制备
合成步骤类似合成聚酰胺1,具体步骤如下:
1)活化:称取图3如式(9)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3中如式(1)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3中如式(8)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,每次进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3中如式(5)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2),对步骤10)的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3中如式(4)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)重复步骤4)、步骤2)的操作,对步骤12)中的相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
14)偶合:称取图3中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
15)重复步骤4)、步骤2)的操作,对步骤14)中的相反应管中的反应物进行盖帽和脱保护反应。为了验证合成产物,进行与步骤4)相应过程相同。
16)偶合:称取图3中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
17)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤16)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
18)偶合:称取图3中如式(7)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
19)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤18)的反应液进行同样的盖帽、脱保护、盖帽反应。为了验证合成产物,进行与步骤4)相应过程相同。
20)偶合:称取图3中如式(5)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
21)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤20)的反应液进行同样的盖帽、脱保护、盖帽反应,得错配聚酰胺的中间体(错配聚酰胺a);为了验证合成产物,进行与步骤4)相应过程相同。
22)将步骤21)的中间体,连有目标聚酰胺2的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤、纯化后,得到淡黄色粉末的目标产物,错配聚酰胺,收率:16.1%。分析hplc:tr=11.97min、98%纯度.1h-nmr(400mhz,dmso-d6):δ10.41(s,1h,芳香的nh),10.36(s,1h,芳香的nh),10.28(s,1h,芳香的nh),10.14(s,2h,芳香的nh),9.95(s,1h,芳香的nh),9.85(s,2h,芳香的nh),9.73(s,2h,芳香的nh),8.28-8.03(m,3h,脂肪的nh),7.87-7.74(m,2h,脂肪的nh),7.63(s,1h,芳香的ch),7.53(s,1h,芳香的ch),7.42(s,1h,芳香的ch),7.30(s,1h,芳香的ch),7.23-7.01(m,6h,芳香的ch),6.88-6.82(m,2h,芳香的ch),6.77-6.63(m,4h,芳香的ch),3.98(s,3h,nch3),3.94(s,3h,nch3),3.89(s,6h,2×nch3),3.84(s,3h,nch3),3.80(s,6h,2×nch3),3.75(s,3h,nch3),3.70(s,6h,2×nch3),3.61(m,2h,nhch2),3.52(m,2h,nhch2),3.48(m,2h,nhch2),3.40-3.25(m,4h,2×nhch2),2.59(m,2h,nme2ch2),2.40(d,3jh,h=7.4hz,2h,coch2),2.30(t,3jh,h=6.8hz,2h,coch2),2.22-2.11(m,4h,2×coch2),2.02(s,6h,n(ch3)2),1.98(s,1h,coch3),1.76(m,2h,ch2ch2ch2),1.53(m,2h,ch2ch2ch2).tof-mass:m/zcalcdforc76h95n30o15[m+h]+:1667.7593;found:1667.6080[m+h]+,1689.8750[m+na]+.
实施例4:吡咯-咪唑聚酰胺类化合物与pd-l1基因启动子区dna相互作用
在体外水平,使用凝胶迁移率实验(emsa)和紫外熔融实验(tm值)考查3种小分子抑制剂(即聚酰胺1、聚酰胺2和错配聚酰胺)与pd-l1基因启动区dna序列相互作用结果,对比所述3种小分子抑制剂之间作用的区别。本发明细胞外研究所应用的dna,包括目标双链dna(wt-ds-dna)、单链dna(ss-dna)、和突变dna(mut-ds-dna),如表2中所示。
表2.本发明中细胞外研究应用的dna
实验方法(一):
(1)电泳样本体系的终体积为10μl,样品中双链dna终浓度为1μm,单链dna终浓度为2μm,dmso含量5%,含100mmtris,1mmedta,50mmnacl。吡咯-咪唑聚酰胺类化合物在样品中的浓度梯度依次为0、1.0、2.0、4.0、8.0、16.0μm。所有样品配置完后,用金属浴在95℃下变性10min,自然冷却至室温后4℃冰箱孵育24h。加入2μl6×loadingbuffer配制成电泳样品,取0.8μl上样。
(2)非变性聚丙烯酰胺凝胶制备
组装凝胶模具,取洁净的小烧杯(50ml)一个,按照表3体系依次加入各种试剂,轻轻混匀,避免气泡产生,快速均匀的将凝胶液注入准备好的电泳玻璃板中,插入梳子,待其完全凝固。
表320%native-page配方
(3)凝胶电泳实验
待凝胶凝固后,拔出梳子,将凝固的凝胶放入电泳槽,内外槽装入1×tbe电泳缓冲液,预电泳30~60min后上样,按预先按设定的电泳条件进行电泳。
电泳条件为:电压150v,温度4.0℃,时间4.5h。
(4)样品染色及成像处理
电泳结束后,将凝胶从玻璃板上剥落,置于聚丙烯酰胺塑料盒内,加入先配好的1×sybrgold核酸染料工作液,避光染色30min后放入凝胶成像仪进行凝胶成像,收集数据,实验结果汇总于图4中。
如图4所示,泳道1:dnamarker(m)。泳道2:单独ss-dna。泳道3:ss-dna+聚酰胺1(16μm)。泳道4:ss-dna+聚酰胺2(16μm)。泳道5:单独wt-ds-dna。图4中,泳道6-10均是表示wt-ds-dna+聚酰胺1,泳道6-10结果所对应的测试样品中聚酰胺1的浓度分别为1,2,4,8或16μm;泳道11-15均是表示wt-ds-dna+聚酰胺2,泳道11-15结果所对应的测试样品中聚酰胺2的浓度分别为1,2,4,8或16μm。泳道16:wt-ds-dna+错配聚酰胺(24μm)。泳道17:单独mut-ds-dna。泳道18:mut-ds-dna+聚酰胺1(16μm)。泳道19:mut-ds-dna+聚酰胺2(16μm)。
实验结果表明:未发现聚酰胺1和2与单链dna序列以及错配聚酰胺与pd-l1基因启动子区dna序列存在显着的结合。当吡咯-咪唑聚酰胺的浓度为1μm时,聚酰胺1和2与pd-l1基因启动子区dna序列(pd-l128nt-wt-ds-dna)存在微弱的结合;当化合物浓度为2μm时,聚酰胺1和2显示出明显的结合条带。当聚酰胺的浓度达到16μm时,双链-dna条带和结合带都被削弱,这可能与beer定律有关,即当化合物的浓度达到一定值时,化合物的高度聚集可能会损害dna。错配聚酰胺与靶标wt-ds-dna几乎没作用;聚酰胺1和2与突变mut-ds-dna几乎没作用。这个发现表明:聚酰胺1和2选择性地与靶标pd-l1启动子序列(wt-ds-dna)浓度依赖性的作用。
实验方法(二):
(1)样本的配制
样本终体积为600μl,样品中目标双链dna(ds-dna)或突变双链-dna(mut-ds-dna)的终浓度为1μm,含有样品溶液中的最终dna浓度为1μm(双链单元),含有100mmtris-hcl,50mmnacl和1mmedta。化合物的终浓度分别为:0、0.5、1、4μm,不含化合物的dna溶液作为对照参考。样品配制结束后,用金属浴在95℃下变性10min,自然冷却至室温后,于4℃冰箱孵育24h。
(2)实验参数的设置
温度范围:1℃→95℃;升温速率:1.0℃/min;测量波长:260nm。
(3)测定:
使用配备的uv-2550分光光度计(shimadzu)测量温度依赖性吸收。以1℃/min的速率将温度从20℃升高至95℃,并测量不同浓度比率下的吸光度值(测定波长:260nm)。测定结束,计算每个样品的熔融温度tm值,三次实验结果平均值±sd结果汇总于表4中。
表4.聚酰胺与目标双链dna(wt-ds-dna)和突变双链-dna(mut-ds-dna)作用的tm值(℃)
实验结果如表4所示,聚酰胺1和2与pd-l1启动子区域匹配的目标双链dna序列相互作用,导致dna的熔融温度升高,而错配聚酰胺导致dna的熔融温度几乎没有变化;聚酰胺1和2对突变双链dna熔融温度几乎没有影响,说明聚酰胺1和2与突变双链dna几乎没有作用。当聚酰胺与dna的摩尔浓度比为1时,聚酰胺1和2使目标双链dna(wt-ds-dna)的熔融温度分别提高了8.2℃和7.0℃,而错配聚酰胺使目标双链dna(wt-ds-dna)基本没有变化。然而,当聚酰胺和dna的浓度比大于1时,熔融温度相比浓度比为1时增加很小,表明当聚酰胺与dna的浓度比为1时,聚酰胺和dna几乎达到饱和状态,说明聚酰胺与靶标dna序列作用是1:1定量关系。对比试验结果表明,聚酰胺1和2可以提高目标双链dna的熔融温度,聚酰胺1更有效,可能是因为它的结构长于聚酰胺2,导致与dna作用的氢键更多,具有更高的细胞外亲和力。错配聚酰胺对目标双链dna的熔融温度没有影响,表明聚酰胺是高度特异性的。
实施例5:聚酰胺与stat3蛋白竞争性结合pd-l1基因启动子区序列
研究发现,stat3能够与pd-l1启动子结合来调控其转录和表达。因此,本发明根据stat3蛋白的结合位点设计化合物,验证吡咯-咪唑聚酰胺与pd-l1启动子序列结合后,加入stat3蛋白以检测聚酰胺竞争结合pd-l1启动子区域中stat3蛋白结合位点的能力。
实施例5中电泳实验的方法重复实施例4中的实验方法(一),不同之处仅在于“某些电泳样品中还加入有stat3蛋白质,stat3蛋白质的终浓度为1μm”。实验结果汇总于图5中。
如图5所示,泳道1:单独wt-ds-dna(1μm)。泳道2:wt-ds-dna+聚酰胺1(12μm)。泳道3:wt-ds-dna+聚酰胺2(12μm)。泳道4:wt-ds-dna+stat3蛋白质(1μm);泳道5-9均是表示wt-ds-dna+stat3蛋白质+聚酰胺1,泳道5-9结果所对应的测试样品中聚酰胺1的浓度分别为1,2,4,8或12μm。泳道10-14均是表示wt-ds-dna+stat3蛋白质+聚酰胺2,泳道10-14结果所对应的测试样品中聚酰胺2的浓度分别为1,2,4,8或12μm。泳道15:wt-ds-dna+错配聚酰胺(24μm)。泳道16:wt-ds-dna+stat3蛋白质+错配聚酰胺(24μm)。双链dna终浓度为1μm。
实验结果显示stat3蛋白与pd-l1启动子区域中的28ntds-dna序列结合,形成两个滞后条带,可能形成两种不同的dna-蛋白质复合物构象。并且随着聚酰胺1或聚酰胺2浓度的增加,与目标双链-dna(wt-ds-dna)结合的stat3蛋白的复合带逐渐减弱,当浓度增加至12μm时,该带几乎消失,表明聚酰胺1或聚酰胺2能竞争性结合到pd-l1启动子区域中stat3蛋白结合位点上。错配聚酰胺对stat3蛋白质与目标wt-ds-dna作用没影响。
实施例6:聚酰胺与pd-l1基因启动子区dna序列结合模式及作用的计算机辅助分子模拟研究
在体外水平,模拟聚酰胺1或聚酰胺2与dna结合的全景图,如图6所示。为简单起见,未表示钠离子和水分子,聚酰胺1或聚酰胺2显示为棒状模型,每条dna链由带状模型表示,构成5'-ttcaccgaa-3'/5'-ttcggtgaa-3'(pd-l1启动子区域中目标双链dna(wt-ds-dna)的作用靶点)复合物的能量最小化结构。而图6中的表格数据显示libdock评估聚酰胺-dna相互作用的结果。
实验方法:使用discoverystudio3.5(accelrys,sandiego,ca)进行分子建模。使用charmm力场参数构建由5'-ttcaccgaa-3'/5'-ttcggtgaa-3'组成的b型dna。基于impypy-γ-pypypy-5'-d(cgctaacaggc)-3'/5'-d(gcctgttagcg)3'复合物的nmr结构构建聚酰胺。周期性边界条件用于显式溶剂模拟。得到的正交水溶剂化模型含有0.145mol/l盐(钠阳离子和氯阴离子),并且距边界的最小距离设定为
实验结果显示模拟和优化结构以获得如图6所示的组合模式。匹配的发夹聚酰胺以反平行的方式与dna小沟中的5'-ttcaccgaa-3'/5'-ttcggtgaa-3'序列紧密结合。我们进行比较与dna序列对接后获得聚酰胺的结合能和libdock评分数据发现,在细胞外实验中,聚酰胺1的对接结果优于聚酰胺2,这与之前的uv熔融实验的结果一致。
实施例7:聚酰胺抑制肿瘤细胞中pd-l1mrna和蛋白的表达
吡咯-咪唑聚酰胺不仅具有特异性,而且具有良好的细胞通透性,易于穿透细胞膜进入细胞核,然后特异性调节靶基因的表达。通过q-pcr实验和免疫印迹实验验证聚酰胺对pd-l1基因的转录和翻译产生影响。本部分使用的主要试剂如表5中。
表5.主要试剂
实验方法(一):将实验细胞复苏,传代培养至生长状态良好,将细胞接种到6孔板中,每孔细胞数为5×105。培养24h,细胞生长至铺满孔板70%~80%左右,分别加入配制好的不同浓度的待测化合物和(或)inf-γ(50ng/ml,一种刺激pd-l1产生的干扰素)。加入待测化合物36h后,观察细胞生长状态及形态,收集细胞提取细胞的总rna。
为了定量pd-l1mrna表达,用trizol试剂(invitrogen)从细胞中提取总rna,并根据firststrandcdnasynthesiskit(toyobo)试剂盒的说明书来合成cdna。使用itaquniversalsybrgreensupermix(biorad)和abiprism7900-ht序列检测系统(96孔,biorad)进行定量实时pcr分析。
(1)qpcr实验所用引物序列委托英潍捷基公司合成。引物序列:pd-l1前引物5'-ggacaagcagtgaccatcaag-3'和后引物5'-cccagaattaccaagtgagtcct-3';gapdh前引物5'-agaaggctggggctcatttg-3'和后引物5'-aggggccatccacagtcttc-3'。其中gapdh为内参对照。
(2)按下列组分配制rt反应液,反应体系10μl(反应体系如表6)。
表6.rt反应体系
rt反应程序:42℃,5分钟;95℃,3分钟;4℃,5分钟。得到的cdna存于4℃备用。
(3)将得到的cdna稀释10倍,按下列组分配制qpcr反应液,反应体系10μl(反应体系如表7)。
表7.qpcr反应体系
(4)将配好的反应液加入到qpcr板中,贴好封板膜,3500rpm混匀,放入qpcr仪中,设置qpcr反应程序:
变性条件:95℃,5分钟;
循环条件:95℃,30秒;60℃,30秒;72℃,30秒;30循环;
延伸条件:72℃,10分钟;
(5)qpcr结束后对所得数据进行分析处理,然后用prism作图。实验重复三次。gapdh作为对照。结果如图7中所示。
实验方法(二):
(1)蛋白提取
将实验细胞复苏,传代培养至生长状态良好,将细胞接种到6孔板中,每孔细胞数为5×105。培养24小时,细胞生长至铺满孔板70%~80%左右,分别加入配制好的不同浓度的化合物和(或)inf-γ(50ng/ml)。加入化合物36h后,观察细胞生长状态及形态,准备提取蛋白。
将6孔板置于冰上,吸取培养液,用预冷的pbs洗涤2~3次。加入1mlpbs,用细胞刮刀将细胞刮下,转移至1.5mlep管中,3500rmp,4℃,离心5分钟。吸出上层pbs液体,加入适量含蛋白酶抑制剂的细胞裂解液,吹打混匀,冰上裂解30分钟。13200rpm,4℃,离心10分钟,回收上清液(细胞总蛋白),用bca法测定蛋白浓度后,于-80℃储存备用。
(2)蛋白定量:按照bca蛋白浓度测定试剂盒说明书,检测样品的蛋白浓度;
(3)电泳:按照表8和表9的配置体系要求配置浓缩胶,分离胶;待胶凝固后,按一定顺序快速上样。先设置电压为80v,当溴酚蓝条带刚好离开浓缩胶进入分离胶后,将电泳电压调整为110v,最终观察到溴酚蓝条带即将跑出分离胶时,电泳结束。
表8.浓缩胶配方
表9.12%sds-page凝胶配方
(4)转膜
取甘氨酸151.1g,tris30.3g,加蒸馏水定容至1000ml,得到10×转膜液。取10×转膜液80ml,加560ml蒸馏水,再加160ml甲醇,配制成800ml1×转膜液,放入冰箱预冷,电泳结束后根据maker的指示条带剥胶,将所需要条带的凝胶切下。将pvdf膜放在甲醇中浸泡30秒,按照阴极-海绵-滤纸-胶-pvdf膜-滤纸-海绵-阳极的顺序组成电转膜的“三明治”结构,在预冷的转膜液中进行,防止气泡的产生。放入转膜槽中,黑色为负极,80v转膜2.5h。
(5)封闭
用tbst配制5%bsa作为封闭液,室摇床封闭2h。
(6)包一抗用tbst按说明书;
(7)用5%bsa将pd-l1单抗和gapdh单抗按说明书中比例稀释,将封闭好的pvdf膜按照蛋白marker指示位置剪膜,加入对应的一抗,4℃摇床过夜。
(8)包二抗,回收一抗,用tbst洗膜3次,每次5分钟,包二抗,室温摇床1~2h,洗膜4次,每次20分钟。
(9)曝光
将ecl化学发光试剂盒的a液与b液按1:1质量比例混匀,加到pvdf膜上,黑暗处理2分钟,将显色好的pvdf膜放到成像仪中拍照记录结果。结果如图8中所示。
图7、8所示,通过荧光定量pcr实验和westernblot实验,证明吡咯-咪唑聚酰胺对hcc827细胞和mda-mb-231细胞中pd-l1基因转录与翻译表达的影响。ifn-γ的刺激能够促进hcc827细胞和mda-mb-231细胞中pd-l1mrna和蛋白的表达,靶向pd-l1基因的聚酰胺1和聚酰胺2对ifn-γ刺激高表达pd-l1的肿瘤细胞hcc827和mda-mb-231中pd-l1mrna的表达(对照图7中分图a和b)和蛋白的表达(对照图8中分图a和b)有不同程度的抑制作用,且抑制作用呈现出浓度依赖的特性;而错配聚酰胺对肿瘤细胞中pd-l1蛋白的表达几乎无影响(对照图8中分图a和b)。聚酰胺1在低浓度时,对hcc827细胞和mda-mb-231细胞的pd-l1mrna和蛋白并没有明显的抑制效果,浓度增加时抑制效果增强;而聚酰胺2对hcc827细胞和mda-mb-231细胞的pd-l1mrna和蛋白的抑制效果在1.25μm时就表现得很明显。聚酰胺对两种肿瘤细胞中pd-l1蛋白表达的影响(图8中a和b)与qpcr检测的结果(图7中a和b)相一致。总的结果表明:聚酰胺2的作用效果更佳,可能是因为聚酰胺对细胞的渗透性与化合物的大小有关,聚酰胺2具有较小的结构,对细胞更具渗透性,并且更容易嵌入dna的小沟中。总之,聚酰胺1和2抑制肿瘤细胞中pd-l1基因的转录和翻译。
实施例8:聚酰胺抑制肿瘤细胞增殖/凋亡和细胞迁移
前面实施例研究发现聚酰胺1和2可以下调肿瘤细胞中pd-l1的表达,本实施例要考察pd-l1的表达下调对肿瘤细胞的凋亡会有什么影响,是通过哪些途径产生的。首先,研究在没有t细胞存在下,聚酰胺是否具有杀伤肿瘤及抑制细胞迁移的作用。本发明研究发现聚酰胺对肿瘤细胞具有直接的杀伤作用,并且抑制细胞迁移,如图9、图10所示。
8.1细胞株:
hcc827(人肺腺癌细胞),mda-mb-231(人乳腺癌细胞)。以上肿瘤细胞株均购自中科院微生物所菌种培养保藏中心。
8.2主要试剂(表10):
表10.主要试剂
实验方法(一):通过使用cck-8实验方法完成聚酰胺对二种肿瘤细胞的细胞毒性研究。
(1)细胞计数:将培养皿中生长状态良好、铺满皿底80%左右的贴壁细胞消化、离心后,用5ml培养基重悬混匀。吸取10μl细胞悬液至细胞计数板,进行计数。计数后用新鲜培养基将hcc827细胞稀释到6×104个/ml,mda-mb-231细胞稀释到8×104个/ml。
(2)铺板:取出96孔板,每孔加入95μl稀释混匀的细胞悬液,剩余4孔加入95μl不含细胞的培养基作为空白对照。然后在96孔板周围一圈加入100μlpbs,以保持孔板微环境的稳定。放入co2培养箱中培养。
(3)化合物的配制与加药:培养至20~24小时左右,孔板中细胞进入对数生长期,按照实验设计的浓度梯度,向对应的实验孔中加入5μl(由1μl化合物溶液和4μl培养基混匀组成,防止化合物局部浓度过大)不同浓度的化合物溶液,使孔中化合物终浓度分别为:2μm、4μm、8μm、16μm、32μm。对照孔只加入dmso,终浓度为1%,空白对照空组不做处理。加药结束后将96孔板放入培养箱中,5%co2,37℃恒温继续培养。
(4)检测:加药处理后的细胞,培养箱中培养48小时后,每孔加入10μlcck-8试剂,放培养箱中保温,每0.5小时用酶标仪检测450nm处的吸光度值。
(5)试验数据处理:按照以下公式计算细胞存活率,然后用prism作图。
细胞存活率(%)=[(as-ab)/(ac-ab)]×100%
as:实验孔(含细胞的培养基、聚酰胺、cck-8)
ac:对照孔(含细胞的培养基、dmso、cck-8)
ab:空白孔(不含细胞的培养基、cck-8)
实验方法(二):通过使用细胞划痕实验(woundhealingassay)方法完成聚酰胺对二种肿瘤细胞迁移的研究。
(1)细胞划痕:将mda-mb-231或hcc827细胞以10×105细胞/ml的细胞密度在dmem或含有生长因子的1640培养基中培养24小时。将培养24h后的细胞用10μl枪头划线,使铺满培养板底部的细胞中间呈现均匀笔直的空隙,移去原培养液,加入1mlpbs洗涤细胞,重复洗涤2~3次,再向孔板中每孔加入1ml不含fbs的培养液,置于显微镜下拍照保存。
(2)加化合物:将拍照完毕的细胞板按每孔设计的化合物浓度加入相应的化合物,充分混匀,放置恒温培养箱中培养48h。
(3)拍照获取实验结果:将加入化合物后继续培养48h的细胞放置电子显微镜下,拍出同一位置加入化合物之后的划痕周围的细胞图片保存。根据原始细胞划痕区距离计算出细胞实际的转移距离。
(4)为了量化实验结果,细胞抑制率(%)通过以下等式计算:细胞抑制率(%)=(1-ddrug/dcontrol)×100%,其中ddrug是药物组中细胞迁移的平均距离,dcontrol是平均值。对照组中细胞迁移的距离,将初始图片与孵育结束时细胞的相应图片进行比较,数据表示为平均值±sd。
实验结果表明:对比图9中a、b、c和表11的结果,可见聚酰胺1和聚酰胺2以浓度依赖方式对hcc827细胞和mda-mb-231细胞增长具有抑制作用;而错配聚酰胺几乎没有作用。同时,聚酰胺对正常细胞mrc-5几乎没有作用。聚酰胺2对hcc827和mda-mb-231细胞的ic50值分别为11.84μm和9.05μm(表11)。综合所得数据,聚酰胺2比聚酰胺1细胞毒性大,极有可能是由于聚酰胺1分子结构大,较聚酰胺2难进入细胞核中与靶点作用。而且聚酰胺1和聚酰胺2对于正常细胞mrc-5几乎没有细胞毒性,说明靶向pd-l1的聚酰胺具有一定的浓度安全域和细胞选择性,为其发展成为靶向药物提供了基础条件。这是在没有t细胞存在的条件下,聚酰胺通过非免疫途径杀伤肿瘤的方式。
如图10所示,聚酰胺1和聚酰胺2以浓度依赖方式对hcc827细胞和mda-mb-231细胞迁移有明显的抑制作用,当聚酰胺1和2的浓度达到1.0μm时,两种细胞的抑制率已达到50%或更高。
表11.吡咯-咪唑聚酰胺抗肿瘤细胞hcc827和mda-mb-231的ic50(μm)值
实施例9:聚酰胺介导t细胞参与的免疫抗肿瘤作用
本发明继续研究了吡咯-咪唑聚酰胺是否可以抑制肿瘤免疫逃逸、通过降低或阻制pd-1/pd-l1之间的作用而激活t细胞免疫功能,从而杀死肿瘤细胞。将t细胞和肿瘤细胞共培养模拟肿瘤微环境,通过elisa实验和cck-8实验检测白介素-2(il-2)因子表达水平和t细胞介导的肿瘤杀伤作用。
实验方法(一):为了分析化合物对肿瘤微环境的影响,将肿瘤细胞与用pma(sigma-aldrich)活化的jurkatt细胞共培养。将共培养物按照5:1(jurkat:肿瘤细胞)的数量比例孵育12或24小时。根据人il-2elisa试剂盒(dakewe)所述测定培养基中分泌的il-2的水平。
实验方法(二):参考上述实施例8实验方法(一),将细胞以每孔8,000个细胞的密度接种到96孔板(corning,3799)中过夜培养,然后暴露于化合物24小时后测试。再向每个孔中加入10μlcck-8(dojindo,ck04)并孵育0.5-1h。使用酶标仪(synergyh1,bio-tek)在450nm下测量每个孔的od值。
实验结果显示:如图11中分图a、b所示,聚酰胺增强了白介素-2(il-2)的表达水平,并且聚酰胺2更有效;和对照组比较,聚酰胺1和2导致hcc827和mda-mb-231细胞中il-2表达的浓度增加约50pg/ml,错配聚酰胺对il-2的表达没有太大影响。用作阳性对照的pd-1/pd-l1抑制剂2(一种由百时美施贵宝(bms)公司开发的、尚未临床应用的小分子pd-1/pd-l1免疫检查点抑制剂)对两种试验细胞影响更明显。这项实验说明,由于聚酰胺1和2下调了肿瘤细胞中的pd-l1表达(实施例7),引起t细胞活化、白介素-2浓度增加,促使免疫t细胞对肿瘤细胞的杀死。在聚酰胺存在下,检测肿瘤细胞的活力结果表明:如图11中分图c、d所示,聚酰胺1和2在共培养体系中对肿瘤细胞具有一定的杀伤作用,导致细胞凋亡,聚酰胺2的效果强于聚酰胺1;错配聚酰胺对肿瘤细胞活力没有太大影响。通过比较图11中分图c、d和图9中分图a、b结果,聚酰胺1和2在同样8μm浓度给药,在jurkatt细胞存在下,增加了hcc827细胞的增长抑制/凋亡率分别为:23.40%和26.14%;对mda-mb-231细胞,分别为:27.76%和30.84%。这些净增加的肿瘤细胞增长抑制/凋亡率是由于聚酰胺通过免疫途径抑制肿瘤的结果。
总之,聚酰胺1和2可以减少免疫逃逸或增强免疫功能,增加il-2的分泌,导致肿瘤细胞凋亡;也可以单独通过非免疫途径抑制肿瘤细胞的增殖和(或)介导肿瘤细胞凋亡。
本说明书所述的内容仅仅是对发明构思实现形式的列举,本发明的保护范围不应当被视为仅限于实施例所陈述的具体形式。
- 还没有人留言评论。精彩留言会获得点赞!