安全性和抗癌活性高的姜黄素和吲哚药物共晶体及其制备方法
本发明涉及姜黄素药物,具体涉及一种姜黄素与吲哚药物共晶体及其制备方法,属于药物化学的技术领域。
背景技术:
姜黄素(curcumin,cur),1,7-双(4-羟基-3-甲氧基苄基)-1,6-庚二烯-3,5-二酮,又称姜黄色素、酸性黄,分子式为c21h20o6,分子量为368.38,熔点约为183℃,粉末呈橙黄色,难溶于水,易溶于丙酮、乙醇、甲醇等有机溶剂,化学结构式如式1。姜黄素是一种天然多酚化合物,从草本植物姜黄、芥末、莪术、郁金等的根茎中提取,属于生物制药分类系统(bcs)第iv类药物,具有低溶解度,低渗透性的特点。几个世纪以来,对其丰富的体外和体内研究报道显示,姜黄素本身具有强大的抗氧化,抗炎,抗菌,抗痉挛,降脂,促进认知等作用,和神经和肝保护作用。世界卫生组织、美国食品药品监督总局等都已经批准把姜黄素应用为食品添加剂,作为一种gras化合物,姜黄素即使在高剂量下也是安全的,即它的剂量可达12000毫克/天,每日允许摄入量为0-3毫克/公斤体重。
姜黄素的巨大的药物治疗价值以及安全性、耐受性引起了人们对使用其治疗各种慢性疾病的兴趣,这些疾病包括癌症、糖尿病、肺结核、心血管、自身免疫性疾病和神经退行性疾病等。然而,姜黄素在高温,强酸,强碱及光照条件下易分解,不稳定,是限制其推广应用的重要原因。同时由于其溶解度低(约11ng/ml)和药代动力学差,导致其生物利用度极其有限,姜黄素的成药及临床应用受到严重阻碍,药效发挥受限。姜黄素的化学结构式如下:
共晶体是两个或多个既可以为分子也可以为离子的物质以一定的化学计量数结合而成的单一晶型物质,既不是盐也不是溶剂化物。基于这一定义,药物共晶体是药物活性成分(activepharmaceuticalingredient,api)和其他共晶形成物(co-crystalcoformer,ccf)按照一定的化学计量比在不改变分子原有的共价结构的同时通过超分子作用力形成的共晶。共晶的形成主要依赖分子间的氢键,π-π双键,范德华作用力等分子间作用力。除了在溶解度、溶解速率、生物有效性和物理稳定性方面有潜在的改进外,药物共晶体还可以增强药物活性组分的其他基本特性,如流动性、化学稳定性、可压缩性和吸湿性等。
在研发固体制剂过程中,口服药物的核心几乎总是晶型固体,共晶是一种提高低水溶性药物的生物利用度的方法。而通过共晶的方式改善姜黄素的性质,相比于其他技术手段,如脂质体、固体分散体、微乳、微球、磷脂复合物等,具有的优势在于:(1)姜黄素作为一种天然药物,具有很多待挖掘的潜在药效,通过寻找安全无毒的共晶形成物与姜黄素相互作用,过程中无需改变药物本身的分子结构,可以最大限度上完全继承姜黄素的药效性质,又达到调控药物的物理化学性质的目的,同时引入的形成物还可与姜黄素发挥协同作用提升其治疗效果。(2)作为晶型药物,由于其结构已知,性质稳定,可以稳定姜黄素的结构,改善其在成药及储存过程中稳定性差的缺点,其他药物剂型往往无法做到,反而容易发生团聚,光不稳定等现象。(3)药物共晶中药物与形成物之间配比确定,有利于用药过程中对于剂量的准确定量,同时无需引入大批量的辅料,可减轻给药剂量。(4)同时,由于结晶工程学和超分子化学的指导,药物共晶相对新药研发具有周期短,投入小,风险低,收益大的优点。而制备脂质体、微球、固体分散体等制剂过程中需要引入大量的载体材料,导致需服用大剂量成药才能达到药效,增加病人负担;同样的,微乳制剂中含有大量的表面活性剂,安全性需慎重考虑;磷脂复合物制备过程中药物需与磷脂在较高温度下进行复合反应,此后去除溶剂,可能导致姜黄素的热降解。因此,要达到制备载药量高,辅体材料少,稳定性好,药效高的姜黄素制剂的目的,药物共晶成为了优秀的技术手段。
目前,中国发明专利cn108840794a公开了一种新型姜黄素药物与针纵酚共晶及其制备方法,中国发明专利cn107721916b中公布了姜黄素-2-氨基吡啶共晶及其制备方法,在不同浓度的乙醇溶液中姜黄素的溶解度得到了改善,但目前大多数姜黄素共晶的形成物仅为一种医药中间体,且毒性大,并无什么大的药用价值,不能真正应用到实际治疗之中。同时采用溶液法进行实验制备可能存在丙酮、二氯甲烷等多种有机溶剂残留,结晶率不高。
学位论文(庞文哲.中药单体姜黄素超分子抗肿瘤复合物的筛选及评价[d].河北医科大学,2018.)中对癌细胞a549的细胞毒性实验表明,姜黄素-哌嗪共晶和姜黄素-异烟碱共晶对癌细胞的抑制率虽然有所提升,但仅比姜黄素高3%-5%,并不能明显提高其抗癌性质,但启示我们通过共晶可提高姜黄素的抗癌毒性。
中国发明专利2007800437308公开了具有与姜黄素类似的结构,且具有抑制aβ聚集的作用、分解聚集aβ的作用、抑制β-分泌酶的作用和保护神经细胞的作用的新型化合物,对治疗老年痴呆有效。该专利启示我们姜黄素的双酮结构和吲哚基团联合作用可拓宽姜黄素的药用范围。但该衍生物是通过有机合成的方法合成具有双酮结构和吲哚基团的化合物,为姜黄素衍生物,且化合物制备的有机合成的方法复杂且需要用到大量有机溶剂,改变了原有物质的化学结构。姜黄素作为一种天然药物具有很大的药用价值,成药过程中一般不期望破坏其原有结构,而药物共晶的方法是在不改变原有物质化学结构的基础上通过分子间作用力使模型药物与共晶形成物相结合,可在最大程度上保留原料药的药用价值。
现有技术文献(singawong,shenyehu,waiwingng,etc.cocrystallizationofcurcuminwithbenzenediolsandbenzenetriolsviarapidsolventremoval[j].crystalgrowth&design,2018,18(9):5534-5546.)采用旋蒸法制备出姜黄素与邻苯二酚、对苯二酚药物共晶。选用多酚类共晶形成物,探索了利用动力学的方法,成功利用如旋蒸法快速成核制备出传统方法难以发现的潜在热力学不稳定的药物共晶,并且所得晶体均表现出较原料药更好的溶出度、吸湿稳定性及压片性能,证明了快速溶剂挥发成核方法制备药物共晶的可行性,但所选共晶形成物并非gras标准的无毒低毒药物。
技术实现要素:
本发明的目的在于针对现有技术中与姜黄素药物形成的共晶体中的原料毒性大,共晶体的抗癌活性有待提高,提供一种共晶原料安全性高,抗癌活性明显增强的安全性和抗癌活性高的姜黄素和吲哚药物共晶体及其制备方法。
本发明针对姜黄素作为一种重要的中药活性成分,但由于水溶性差,人体难以吸收,临床应用受到限制。发现吲哚与姜黄素形成的共晶不但可提高姜黄素药物的溶解度和溶出速率,而且对药物的抗癌活性有相比现有的姜黄素共晶体有显著的增强,可能是姜黄素与吲哚的共晶形成物产生分子间相互作用后,共晶形成物首先快速溶解,从晶格析出,从而带动药物的溶出,此时溶解度会大大提升;尤其是吲哚相比于现有的姜黄素共晶原料安全性有保证,克服了现有技术的共晶原料的安全性问题。
吲哚(indole,ind)又称苯并吡咯,包含了一个六元苯环和一个五元含氮的吡咯环,是一个芳香杂环有机化合物,分子式为c8h7n,分子量为117.15,化学结构式如式2。吲哚及其同系物和衍生物是重要的有机原料和精细化工产品,广泛存在于自然界,主要存在于天然花油,如茉莉花、甜橙、白柠檬、柚皮、柑橘、苦橙花、水仙花、香罗兰等中,主要用作香料、染料、氨基酸的原料。吲哚为允许使用的食用香精,符合美国gras及欧洲fema规定的食品添加剂范畴,天然无危害的食品添加剂属性使其作为药用辅料的应用大大推广,不会增加药物制剂的生物毒性。
本发明针对生物药剂学分类系统bcsiv类药物姜黄素,存在的低溶解性、稳定性差、抗癌作用受限的问题,通过设计药物共晶以改善姜黄素的理化及生化性质,挑选美国食品药品监督总局规定的gras类天然药物吲哚作为共晶形成物,设计得到的共晶体具有更好的溶解性及溶出度;共晶体对于癌细胞的细胞毒性研究表明,该共晶体能更有效地抑制癌细胞的生长,吲哚作为一种安全的药物活性成分的加入,保证了药物共晶体的药用安全性,可协同姜黄素达到更好的抗肿瘤效果。同时具有良好的储存稳定性及吸湿性能,晶型稳定,有效避免了现有晶型转变造成的性质改变问题;
本发明目的通过以下技术方案实现,具有如下特征:
安全性和抗癌活性高的姜黄素和吲哚药物共晶体,由姜黄素和吲哚按摩尔比为1:1或1:2或1:3混合均匀后加入有机溶剂,溶剂蒸发后析出姜黄素和吲哚药物共晶体;
当姜黄素与吲哚摩尔比为1:1,所得共晶体中姜黄素、吲哚及姜黄素和吲哚共晶同时存在;用cu-kα辐射,其以2θ表示的x射线衍射谱图在5.70±0.2°,6.22±0.2°,7.08±0.2°,10.18±0.2°,11.36±0.2°,15.18±0.2°,17.34±0.2°,37.04±0.2°处有特征峰;
当姜黄素与吲哚摩尔比为1:2,所得共晶体为纯姜黄素和吲哚共晶;用cu-kα辐射,其以2θ表示的x射线衍射谱图在5.70±0.2°,6.22±0.2°,7.08±0.2°,10.18±0.2°,11.36±0.2°,15.18±0.2°,17.34±0.2°处有特征峰;
当姜黄素与吲哚摩尔比为1:3,所得共晶体为吲哚、姜黄素和吲哚共晶共存;用cu-kα辐射,其以2θ表示的x射线衍射谱图在5.70±0.2°,6.22±0.2°,7.08±0.2°,10.18±0.2°,11.36±0.2°,15.18±0.2°,17.34±0.2°处有特征峰。
为进一步实现本发明目的,优选地,使用差式扫描量热法测定dsc曲线进行测试时,存在如下三种情况:
当姜黄素与吲哚摩尔比为1:1,所得姜黄素和吲哚药物共晶体存在三个吸热峰,对应的温度范围是50.28~56.04℃,其峰值为51.00℃;69.30~83.24℃,其峰值为79.22℃;109.5~170.14℃,其峰值为161.60℃;
当姜黄素与吲哚摩尔比为1:2,所得姜黄素和吲哚药物共晶体存在两个吸热峰,对应的温度范围是77.19~87.24℃,其峰值为82.02℃;121.31~150.30℃,其峰值为145.67℃;
当姜黄素与吲哚摩尔比为1:3,所得姜黄素和吲哚药物共晶体存在三个吸热峰,对应的温度范围是50.28~56.04℃,其峰值为50.99℃;77.19~87.24℃,其峰值为81.97℃;119.31~153.30℃,其峰值为143.57℃。
优选地,在ph=1.5的模拟人工胃液中,姜黄素溶解度为0.21微克/毫升,姜黄素和吲哚药物共晶体的溶解度为0.67-0.88微克/毫升。
所述的安全性和抗癌活性高的姜黄素和吲哚药物共晶体的制备方法:将姜黄素和吲哚按摩尔比为1:1或1:2或1:3混合均匀,在混合均匀的姜黄素和吲哚药物原料中加入有机溶剂中,超声使其充分溶解混匀,得到黄色透明溶液,加热使溶剂挥发完全,晶体析出后得产物。
优选地,所述的有机溶剂为丙酮、乙醇、甲醇中的一种或两种及以上的混合。
优选地,每毫克姜黄素加入0.5~0.8毫升的有机溶剂。
优选地,所述的加热使溶剂挥发是在真空旋蒸挥发器中进行,水浴加热。
优选地,所述的真空旋蒸挥发器的旋蒸时间为30~90min,水浴加热的温度为40~70℃,真空度为-0.06~-0.1mpa,旋转速度为100~200r/min。
优选地,所述的混合均匀是指将姜黄素和吲哚加入离心管中,于旋涡混合器中涡旋混合至完全混合均匀。
相比于已有成果及技术手段,本发明具有以下优势:
(1)本发明选用gras标准的安全天然药物成分吲哚作为共晶形成物与模型药物姜黄素形成药物共晶,作为食品添加剂,保证了药物的安全性,相比于其他医药中间体,药物毒性低,无副作用。
(2)本发明公开的姜黄素和吲哚共晶克服了药物制备过程中重现性差的问题,同时溶解度,溶出度相对于原料药姜黄素均有所提高。
(3)本发明公开的姜黄素和吲哚共晶体稳定性吸湿性良好,避免了固态药物储存及运输过程中易发生晶型转变,药物分解的问题。
(4)本发明公开的姜黄素和吲哚共晶体体外抗肿瘤实验表明该共晶体能使癌细胞活率降低,相较于原料药能更好的抑制癌细胞。
(5)本发明公开的姜黄素和吲哚共晶体通过与共晶形成物形成共晶体的形式,形成了新的稳定晶型,避免了单一的药物晶型发生晶型转变产生的晶型不稳定问题。
(6)本发明的技术方案的制备方法操作简便,本发明的原料及所用试剂均市售可得,且毒性低,环境污染小,产品收率高。
附图说明
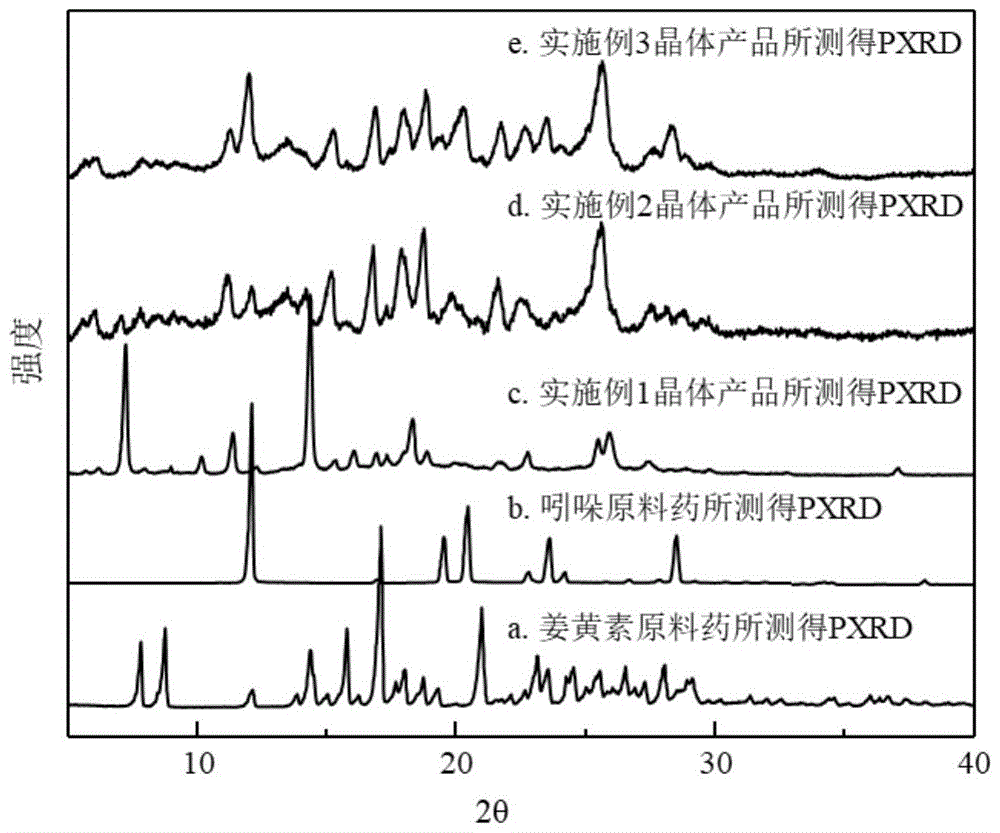
图1是姜黄素、吲哚原料药及姜黄素-吲哚药物共晶体实验测得的pxrd图谱。
图2是姜黄素-吲哚药物共晶体实验测得的dsc图谱。
图3是姜黄素、吲哚原料药、姜黄素和吲哚物理混合物,及姜黄素-吲哚药物共晶体实验测得的dsc图谱。
图4是姜黄素、吲哚原料药、姜黄素和吲哚物理混合物,及姜黄素-吲哚药物共晶体实验测得的ft-ir图谱。
图5是姜黄素、姜黄素-吲哚药物共晶体在ph=1.5(0.4%十二烷基硫酸钠)的人工胃液中姜黄素的释放曲线。
图6是姜黄素原料药、姜黄素-吲哚药物共晶体在室温,相对湿度为80%的密闭环境内避光放置一个月时间前后实验测得的pxrd图谱。
图7是吲哚药物对表皮细胞hacat的细胞毒性实验结果图。
图8是姜黄素、吲哚原料药、及姜黄素-吲哚药物共晶体实验测得的分别对a549癌细胞的体外毒性实验结果图。
具体实施方式
为更好地理解本发明,下面结合附图和实施例对本发明作进一步的说明,但是本发明的实施方式不限如此。
实施例1
姜黄素-吲哚药物共晶的制备:用电子天平准确称量摩尔比为1:1的姜黄素100mg和吲哚31.8mg,使用涡旋混合器混合均匀,加入30ml丙酮,超声使药物完全溶解并混合均匀,转移于圆底烧瓶内,使用真空旋蒸蒸发仪在水浴温度40℃,旋转速率200r/min,真空的条件下旋转蒸发使溶剂完全去除,随后放置于40℃真空干燥箱中干燥过夜,使溶剂彻底挥发完全,得到橙红色固体粉末状产品。
对实施例1得到的姜黄素和吲哚共晶进行x射线粉末衍射(pxrd)表征,仪器为德国brukerd8advance,得到的pxrd图谱如附图1中所示,其中a是姜黄素原料药的pxrd图谱,b是吲哚原料药的pxrd图谱,c为实施例1所得晶体产品测得的pxrd图谱,对比于两种原料药谱图a和b,可见所得产品谱图c中呈现的a,b谱图的特征峰减弱,而且在5.70±0.2°,6.22±0.2°,7.08±0.2°,10.18±0.2°,11.36±0.2°,15.18±0.2°,17.34±0.2°,37.04±0.2°处出现了较强的新的特征峰,证明得到的产品为新的晶型物质。
对实施例1姜黄素和吲哚药物共晶进行差热扫描量热分析。附图2是通过tadsc250差示扫描量热仪进行测定,氮气氛围,升温速率为10℃/min的产品dsc图谱。对实施例1得到的姜黄素和吲哚共晶进行dsc谱图分析,存在三个吸热峰,对应的温度范围是50.28~56.04℃,其峰值为51.00℃;69.30~83.24℃,其峰值为79.22℃;109.5~170.14℃,其峰值为161.60℃。
附图3所示的dsc谱图,图中a和b分别代表姜黄素原药和吲哚原药,c为两种原药物理混合,d为本发明实施例1制备的姜黄素和吲哚共晶。可见,姜黄素的熔融吸热峰在186.35℃,吲哚的熔融吸热峰在53.13℃,物理混合的熔融吸热峰为姜黄素和吲哚熔融吸热峰的叠加,在53.13℃、156.96℃;而共晶在79.54℃出现了新的区别于两个原料药及物理混合物的熔融吸热峰。从a,b,c,d的谱图差异也可证明有新相的生成,并且制备得到的新共晶的熔点小于姜黄素的熔点,略大于吲哚熔点值,处于两种原料药熔点之间,排除了低共熔混合物生成的可能性,形成了药物共晶。
对实施例1得到的姜黄素和吲哚药物共晶进行红外光谱(ft-ir)分析,测试仪器为德国brukertensor27,得到如附图4所示的红外吸收光谱。图中a和b分别代表姜黄素原料药和吲哚原料药,c为两种原料药的物理混合物,d为实施例1制备的姜黄素和吲哚药物共晶。姜黄素图谱中3501cm–1为酚羟基的振动吸收峰,1631cm–1为c=o双键的振动吸收峰,1601cm–1为苯环c=c双键的振动吸收峰;吲哚图谱3405cm–1是n-h的振动吸收峰,1414cm–1左右是c-n的伸缩振动峰。物理混合图谱为两种原料药的混合物,共晶图谱3381cm–1左右为酚羟基和n-h的振动吸收峰,1626cm–1为c=o双键的振动吸收峰,1582cm–1为苯环c=c双键的振动吸收峰;共晶中特征峰发生偏移,说明由于氢键的作用形成了共晶。
通过以上xrd、dsc、ft-ir综合分析,成功制备了姜黄素-吲哚药物共晶。
对实施例1进行溶解度测试,测定方法如下:(1)取过量的100毫克实施例1的姜黄素和吲哚共晶体样品于100ml的棕色瓶中加入20ml的溶液;(2)将棕色瓶放入提前设定温度为37℃,转速为100rpm的恒温振荡箱内;(3)振荡18h后,取2ml溶液经过0.22μm的滤膜过滤后采用紫外可见分光光度计进行定量。实验测得在含20%(w/w)乙醇-水溶液中,姜黄素原料药的溶解度为0.2628微克/毫升,实施例1产品的溶解度为0.9181微克/毫升,约为原料药的3.5倍;在ph=1.5的人工胃液(0.2%sds)中,姜黄素溶解度为0.21微克/毫升,实施例1产品的溶解度为0.78微克/毫升,约为原料药的3倍;在ph=7.4的pbs缓冲溶液中姜黄素的溶解度为0.025微克/毫升,实施例1产品的溶解度为0.76微克/毫升,约为原料药的30倍。表明药物共晶明显改善了姜黄素的溶解度。
对实施例1进行溶出度测试,测定方法如下:称取0.015毫克的实施例1所得的姜黄素和吲哚共晶体产品放入装有40ml的含0.4%(w/w)十二烷基硫酸钠的人工胃液(ph=1.5)的离心管内,并将离心管放在预先设定的转速为100rpm,温度为37℃的恒温振荡箱内,随后,在固定的时间间隔内将离心管放置于离心机中,在9000r条件下离心5min,从容器的上清液中取出1ml释放介质,并补充1ml缓冲溶液,用0.22μm的滤膜过滤后,用紫外可见分光光度计对姜黄素的含量进行定量分析。结果见附图5,图中a为姜黄素原料药的溶出曲线,b为实施例1所得晶体产品的溶出曲线,可见共晶产品的溶出速率明显提升,较原料药提升30%左右,共晶可以改善姜黄素的溶出速率。
对实施例1进行稳定性测试,测定方法如下:称取100毫克共晶产品在室温,相对湿度为80%的密闭环境内避光放置一个月,通过pxrd测试结果对比样品前后的晶型的变化,见附图6,a为实施例1稳定性实验前的pxrd图谱,b为实施例1稳定性测试一个月后的pxrd图谱,可见共晶产品晶型稳定,不易发生晶型转变。
选用人体表皮细胞hacat对所选用的共晶形成物吲哚进行药物安全性评价,及选用非小细胞肺癌细胞a549对实施例1进行体外细胞毒性测试,测定方法如下:选用人体表皮细胞hacat及非小细胞肺癌细胞a549细胞,取复苏好的细胞置于含10%胎牛血清的dmem培养基中、放于37℃、含有5%co2的恒温恒湿箱中培养。实验采用cck8法研究姜黄素原料药、吲哚共晶形成物及姜黄素和吲哚药物共晶的细胞毒性。将培养皿中的细胞充分清洗消化,并用含胎牛血清培养基稀释成5×104个/ml的细胞悬浮液,按每个孔含5000个细胞将细胞均匀铺在96孔板的孔内,每次吸取细胞需吹打均匀,尽量保证吸取细胞数量相同,然后放于37℃、含有5%co2的恒温恒湿箱中培养24小时。24小时后,将孔板中的培养基去除,加入不含血清的培养基饥饿细胞2小时后,加入配置好的含有一定浓度一定浓度梯度的姜黄素、吲哚、姜黄素-吲哚共晶药物的0.8%胎牛血清培养基50微升,在空白孔中及在对照孔中,加入100微升含0.4%胎牛血清的培养基,空白孔和对照孔均设置6个复孔。给药后培养24小时,载药颗粒的药物浓度与姜黄素的药物浓度相等,每个实验孔均设置3个复孔。24小时后,在每个孔中加入10微升cck8试剂,避光培养2小时,用酶标仪检测每个孔在630nm和430nm处的吸光度差值,扣除空白孔与对照孔的吸光值后即可算出每个孔的细胞活率。
药物安全性实验结果见附图7,图为各浓度下吲哚对人体表皮细胞hacat的细胞活率。可见在药物浓度梯度内所选的共晶形成物吲哚对人体表皮细胞的细胞活率均在90%以上,表明吲哚对人体正常细胞并无毒性,根据gras及fema规定,是可以使用的安全性药物或食品添加剂,ld50为1000毫克/千克。
细胞毒性实验结果见附图8。各药物浓度梯度分别为:浓度1:姜黄素(100微克/毫升)、吲哚(63.6微克/毫升);浓度2:姜黄素(50微克/毫升)、吲哚(31.8微克/毫升);浓度3:姜黄素(25微克/毫升)、吲哚(15.9微克/毫升);浓度4:姜黄素(12.5微克/毫升)、吲哚(7.95微克/毫升);浓度5:姜黄素(6.125微克/毫升)、吲哚(3.975微克/毫升);姜黄素和吲哚药物共晶体中所含姜黄素和吲哚含量与原料药相同。由图8可知,即便在最高浓度1时,姜黄素原料药和吲哚原料药对癌细胞a549作用的细胞活率均在80%以上,且随着药物浓度降低,细胞毒性逐渐减弱,而在浓度1时,姜黄素和吲哚药物共晶体对a549作用的细胞活率下降到20%左右,相比于原料药极大地加强了对肺癌细胞的抑制作用,相同药物浓度下,细胞活率仅为原料药的1/4,且随着浓度降低,细胞毒性均呈现出高于原料药的现象。这可能是由于姜黄素和吲哚药物共晶体的形成增加了药物的溶解度和溶出速率,使药物更多更快的被细胞吸收,从而使其对癌细胞具有更好的抑制效果。
综上所述,该姜黄素和吲哚共晶体在增强溶解度、溶出度、稳定性的同时也增强了药物抗肿瘤活性,解决了姜黄素的生物利用度差的缺陷,大大提高了姜黄素对于癌细胞的抑制作用,使得该共晶体在肺癌化疗方案中具有更广泛的应用。
实施例2
姜黄素-吲哚药物共晶的制备:用电子天平准确称量摩尔比为1:2的姜黄素100mg和吲哚63.6mg,使用涡旋混合器混合均匀,加入30ml丙酮,超声使药物完全溶解并混合均匀,转移于圆底烧瓶内,使用真空旋蒸蒸发仪在水浴温度40℃,旋转速率200r/min,真空的条件下旋转蒸发使溶剂完全去除,随后放置于40℃真空干燥箱中干燥过夜,使溶剂彻底挥发完全,得到橙红色固体粉末状产品。
图1所示的pxrd谱图、图3所示的dsc谱图特征及图4所示的红外光谱图特征,说明本实施例所得固体产品为姜黄素和吲哚共晶。
对实施例2进行溶解度测试,结果与实施例1相近。实验测得在含20%(w/w)乙醇-水溶液中,实施例2产品的溶解度为0.9881微克/毫升,在ph=1.5的人工胃液(0.2%sds)中,实施例2产品的溶解度为0.88微克/毫升,约为原料药的3倍;在ph=7.4的pbs缓冲溶液中实施例2产品的溶解度为0.79微克/毫升,约为原料药的30倍。
对实施例2进行溶出度测试,结果见附图5,曲线c为实施例2的溶出度曲线。可见实施例2的溶出速率明显提升,提高了20%,共晶可以改善姜黄素的溶出速率。
对实施例2进行稳定性测试,测定方法如下:称取100毫克共晶产品在室温,相对湿度为80%的密闭环境内避光放置一个月,通过pxrd测试结果对比样品前后的晶型的变化,见附图6,c为实施例2稳定性实验前的pxrd图谱,d为实施例2稳定性测试一个月后的pxrd图谱,可见共晶产品晶型稳定,不易发生晶型转变。
对实施例2进行体外细胞毒性测试,实验结果见附图8,可知,姜黄素原料药和吲哚原料药对癌细胞a549作用的细胞活率均在80%以上,而在浓度1时,实施例2对a549作用的细胞活率下降到18%,与实施例1结果相似,姜黄素-吲哚共晶使姜黄素对癌细胞具有更好的抑制效果。
实施例3
姜黄素-吲哚药物共晶的制备:用电子天平准确称量摩尔比为1:3的姜黄素100mg和吲哚95.4mg,使用涡旋混合器混合均匀,加入30ml丙酮,超声使药物完全溶解并混合均匀,转移于圆底烧瓶内,使用真空旋蒸蒸发仪在水浴温度40℃,旋转速率200r/min,真空的条件下旋转蒸发使溶剂完全去除,随后放置于40℃真空干燥箱中干燥过夜,使溶剂彻底挥发完全,得到固体粉末状产品,即为姜黄素和吲哚药物共晶。
图1所示的pxrd谱图、图3所示的dsc谱图、图4所示的红外光谱特征,均说明本实施例所得固体产品为姜黄素和吲哚药物共晶。
对实施例3进行溶解度测试,结果与实施例1例2相近。实验测得在含20%(w/w)乙醇-水溶液中,实施例3产品的溶解度为0.7898微克/毫升,在ph=1.5的人工胃液(0.2%sds)中,实施例3产品的溶解度为0.67微克/毫升,约为原料药的3倍;在ph=7.4的pbs缓冲溶液中实施例3产品的溶解度为0.69微克/毫升,约为原料药的30倍。
对实施例3进行溶出度测试,结果见附图5,曲线d为实施例3的溶出度曲线。可见实施例3的溶出速率明显提升,提高了20%,共晶可以改善姜黄素的溶出速率。
对实施例3进行稳定性测试,测定方法如下:称取100毫克共晶产品在室温,相对湿度为80%的密闭环境内避光放置一个月,通过pxrd测试结果对比样品前后的晶型的变化,见附图6,e为实施例3稳定性实验前的pxrd图谱,f为实施例3稳定性测试一个月后的pxrd图谱,可见共晶产品晶型稳定,不易发生晶型转变。
对实施例3进行体外细胞毒性测试,实验结果见附图8,可知与实施例1例2结果相似,姜黄素-吲哚共晶使姜黄素对癌细胞具有明显更好的抑制效果。
综上,姜黄素是一种重要的多酚类植物提取物,具有抗肿瘤、抗炎、抗氧化等多种药理活性。在1985年,印度科学家首次提出姜黄素具有抗癌作用,但其溶解度低、渗透性低,尤其是在水中的溶解度极低,导致其生物利用度低,临床应用受到极大的限制;目前现有技术也选择通过共晶来解决原药存在是问题,但是现有技术研究者们大多选择酰胺类、对苯二酚、间苯二酚、邻苯三酚等非天然化合物作为形成物;或仅为一种医药中间体,如针纵酚、4-氨基苯酚、4-羟基吡啶等,但这些物质毒性较大,对人体无益。由于这些化学合成化合物的毒副作用,使共晶研究受到一定的限制,与姜黄素形成共晶后即使性能略有提升也无法真正应用于药物制剂,无法应用于临床癌症治疗。
本发明姜黄素共晶引入的共晶形成物吲哚是安全的。附图7实验也可证明其药用安全性,在药物浓度梯度内所选的共晶形成物吲哚对人体表皮细胞的细胞活率均在90%以上,表明吲哚对人体正常细胞并无毒性,根据gras及fema规定,是可以使用的安全性药物或食品添加剂,ld50为1000毫克/千克,不存在引入有毒物质而需考虑药物毒性的问题,对扩大姜黄素的临床应用具有很大价值。尤其是,本发明由于共晶物溶解度、渗透和稳定性的提高,使得抗癌活性也有显著的提升。由图8可知,即便在100μg/ml时,姜黄素原料药和吲哚原料药对癌细胞a549作用的细胞活率均在80%以上,且随着药物浓度降低,细胞毒性逐渐减弱,而姜黄素和吲哚药物共晶体对a549作用的细胞活率下降到20%左右,细胞活率仅为原料药的1/4,且随着浓度降低,细胞毒性均呈现出高于原料药的现象。即使在最低浓度6微克/毫升时,姜黄素作用下的癌细胞几乎并无死亡现象,而姜黄素-吲哚共晶3个实施例中的细胞存活率仅为85%左右,说明即使在低浓度下姜黄素-吲哚共晶也能发挥有效的抗癌作用。相比于现有技术中效果相对较好的姜黄素-哌嗪共晶,姜黄素-异烟碱共晶,本发明在相同的姜黄素浓度下,抗癌活性有明显的提升,尤其是在高浓度25微克/毫升时,抗癌活性提升了30%-40%,这极大提高了共晶物的药物应用价值,即使是在较低浓度的6微克/毫升时仍有10%以上的提升。可能是姜黄素-吲哚共晶可提高姜黄素的体内吸收,从而使其对癌细胞具有更好的抑制效果。具体测试结果见表1。
表1不同姜黄素浓度下各共晶相较于原料药的细胞抑制提升百分率
总体来说,本发明中原料药姜黄素、吲哚毒性小,所以即便高浓度用药也具备安全性。姜黄素的剂量可达12000毫克/天,每日允许摄入量可达3毫克/公斤体重;吲哚为1000毫克/千克体重,即使在高剂量下也是安全的。而且本发明中姜黄素-吲哚共晶抗癌药效增强,且无需引入大量辅料,在疾病治疗过程中,用药剂量和给药次数可减少,减轻病人负担及压力。通过姜黄素与吲哚共晶就可解决姜黄素原料具有人体难以吸收,导致其药效低下、生物利用度低的问题。在100微克/毫升时,姜黄素-吲哚共晶的癌细胞活率仅为20%,且随着浓度的增加,活率会越来越低,达到几乎完全抑制的效果。同时本发明姜黄素-吲哚共晶抗肿瘤作用提升,可在化疗治疗晚期癌症阶段显示出好的协同作用,且克服化疗中最大的障碍,多药抗药性,其使用受限制的主要因素是化疗药的毒副作用较大,患者无法耐受,而本发明药物安全性好,对患者身体危害降低。
- 还没有人留言评论。精彩留言会获得点赞!