基于给体-受体结构的四齿环金属铂(II)或钯(II)配合物发光材料及其应用的制作方法
本发明涉及一种发光材料及其应用,尤其涉及一类具有延迟荧光性质的四齿环金属铂(ii)或钯(ii)配合物发光材料及其在有机发光元件中的应用。
背景技术:
有机发光二极管(oled)具有分辨率高、响应快、色彩鲜艳、制造成本低等优点,因此被认为是最有潜力的新一代显示和照明技术,同时也受到学术界和工业界的广泛关注,具有广阔的应用前景。
发光材料是oled器件的核心材料,也是此领域发展的关键,但是具有稳定高效且能够满足商业化应用的发光材料依然极其稀少,尤其是量子效率高的磷光和延迟荧光材料。因此,新型高性能发光材料的设计发展依然是促进oled领域发展的重要研究方向。此外,oled器件的稳定性是其商业化应用的前提,而提高发光材料分子的辐射速率可以使分子高效辐射发光;同时缩短分子处于激发态的时间,减小分子激发态寿命τobs,减少甚至避免发生三线态-三线态激子湮灭而产生高能量的单线态激子;此外,减少激发态分子非辐射弛豫产生的热能,提高材料分子和oled器件的稳定性。因此,提高发光材料的辐射速率kr可缩短其激发态寿命τobs,对于提高材料分子和oled器件的稳定性具有重要意义。
技术实现要素:
为了提高辐射速率krobs以设计发展具有短激发态寿命τobs的发光材料,本发明提供了一类基于给体-受体结构的四齿环金属铂(ii)或钯(ii)配合物发光材料,具有通式(1)所示结构:
其中,
m独立地表示为金属铂(ii)或者钯(ii);
ra、rs、rc、rd和re各自独立地表示单取代、双取代、三取代、四取代或者无取代,且ra、rs、rc、rd和re各自独立地为氢、氘、烷基、烷氧基、环烷基、杂环基、烯基、取代或未取代的芳基、取代或未取代的芳氧基、取代或未取代的杂芳基、卤素、羟基、巯基、硝基、氰基、氨基、羧基、磺基、肼基、脲基、炔氧基、酯基、酰胺基、磺酰基、亚磺酰基、磺酰基胺基、磷酰基胺基、烷氧基羰基胺基、芳氧基羰基胺基、甲硅烷基、烷基胺基、双烷基胺基、单芳基胺基、双芳基胺基、亚脲基、亚胺基或其组合;且两个或者多个邻近的ra、rb、rc、rd和re可选择性连接形成稠环;
给体d1或d2各自独立地选择为以下结构:
其中,r1、r2、r3、r4、r7、r8、r10、r11、r12、r13、r14和r15各自独立地选择为氢、氘、卤素、取代或者未取代的烷基、取代或者未取代的烷氧基、取代或者未取代的环烷基、取代或者未取代的杂环基、取代或者未取代的芳基、取代或者未取代的芳氧基、取代或者未取代的硅基、取代或者未取代的单或二c1-c24烷基氨基、取代或者未取代的单或二芳基氨基、氰基或其组合;其中相邻的取代基可以稠合成环;
o1、p1、q1、r1、s1、t1、u1、v1、w1、x1、y1和z1分别为r1、r2、r3、r4、r7、r8、r10、r11、r12、r13、r14和r15的个数;
o1和p1是0-5的整数;q1、r1、s1、t1、u1、v1、w1、x1、y1和z1是0-4的整数;
进一步地,本发明所保护的通式(1)可以优选出以下具体结构化合物化1~化65这些化合物仅为代表性的,m表示为金属铂(ii)或者钯(ii):
本发明的目的之二,是提供一种有机电致发光器件。有机电致发光器件包括阳极、阴极以及位于所述阳极和阴极之间的至少一层的有机薄膜,有机薄膜中含有通式(1)所表示的一种或者多种有机电致发光化合物。所述有机层包括发光层和功能层,通式(1)所表示的化合物可以作为发光材料单独或者掺杂混合使用。
本发明的有益效果包括:
通过给体-受体结构可以调控材料分子激发态性质,减小其最低单线态激发态(s1)和最低单线态激发态(t1)之间的能级δest,提高分子的系间窜越速率,进而提高辐射速率kr和缩短激发态寿命τobs;同时提高材料分子的磷光量子效率。
附图说明
此处所说明的附图用来提供对本申请的进一步理解,构成本申请的一部分,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。在附图中:
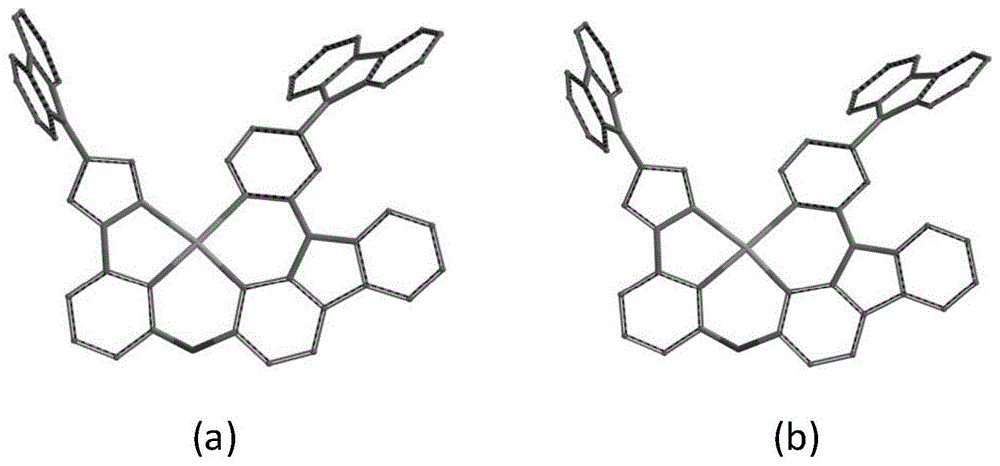
图1为通过密度泛函理论(dft)计算得到的pt1和pd1的优化后的分子结构;
图2为pt1、pd1和其配体在室温[300k]下二氯甲烷溶液中的吸收光谱比较,ligand代表配体;
图3为pt1在77k和室温[300k]环境下的发射光谱图比较,其中2-methf为2-甲基四氢呋喃,dcm为二氯甲烷,pmma为聚甲基丙烯酸甲酯;
图4为pd1在77k和室温[300k]环境下的发射光谱图比较,其中2-methf为2-甲基四氢呋喃,dcm为二氯甲烷,pmma为聚甲基丙烯酸甲酯;
图5为pt1的光稳定性测试曲线;
图6为有机发光元件的结构示意图,图中从下到上依次表示衬底、阳极、空穴注入层、空穴传输层、发光层、电子传输层和阴极。
具体实施方式
下面将以多个合成实施例为例来进一步说明本发明的原理和特征,所举的实施例只用于解释本发明,但并非用于限定本发明的范围。
下面举例说明下述通式(1)代表的本发明的磷光发光材料的具体实例,然而,不解释为限制本发明。
除非另有说明,以下试验中所涉及到的所有商业试剂购买后直接使用,没有进一步纯化。核磁共振氢谱和碳谱均在氘代氯仿(cdcl3)或氘代二甲基亚砜(dmso-d6)溶液中测得,氢谱使用400或500兆赫兹的核磁共振谱仪,碳谱使用100或126兆赫兹的核磁共振谱仪,化学位移以四甲基硅烷(tms)或残留溶剂为基准。如果用cdcl3作溶剂,则氢谱和碳谱分别以tms(δ=0.00ppm)和cdcl3(δ=77.00ppm)作为内标。如果用dmso-d6作溶剂,则氢谱和碳谱分别以tms(δ=0.00ppm)和dmso-d6(δ=39.52ppm)作为内标。以下缩写(或组合)用于解释氢谱峰:s=单峰,d=双重峰,t=三重峰,q=四重峰,p=五重峰,m=多重峰,br=宽峰。高分辨质谱在appliedbiosystems公司的esi-qtof质谱仪上测得,样品电离模式为电喷雾电离。
合成实施例1:四齿环金属铂(ii)配合物磷光发光材料pt1合成路线
中间体3的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入化合物1(325mg,1.00mmol,1.0当量),化合物2(390mg,1.20mmol,1.2当量),碘化亚铜(10mg,0.05mmol,5mol%),2-甲酸吡啶(12mg,0.10mmol,10mol%),磷酸钾(425mg,2.00mmol,2.0当量),然后抽换氮气三次,在氮气保护下加入二甲基亚砜(10ml)。该混合物在90℃油浴中搅拌反应2天,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的盐水,并用乙酸乙酯萃取。有机层用水洗涤两次,水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,洗脱剂:石油醚/乙酸乙酯=10:1-5:1,得到中间体3,粘稠液体438mg,收率84%。1hnmr(500mhz,cdcl3):δ7.04-7.06(m,1h),7.13(d,j=8.5hz,2h),7.30(t,j=2.0hz,1h),7.32(s,1h),7.34(s,1h),7.44-7.50(m,8h),7.54-7.56(m,2h),7.60-7.63(m,1h),7.85(dd,j=8.0,1.5hz,1h),8.01(s,1h),8.13(d,j=7.5hz,2h),8.25(d,j=0.5hz,1h)。
中间体4的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入化合物3(280mg,0.54mmol,1.0当量),三苯基膦(422mg,1.61mmol,3.0当量),然后抽换氮气三次,在氮气保护下加入邻二氯苯(10ml)。该混合物在180℃油浴中搅拌反应24小时,薄层色谱监测至原料反应完毕,冷却至室温。减压蒸馏除去溶剂,所得粗品用硅胶层析柱分离提纯,洗脱剂:石油醚/二氯甲烷=10:1-5:1,得到中间体4,白色固体218mg,收率82%。1hnmr(500mhz,cdcl3):δ7.01-7.05(m,2h),7.15(d,j=2.0hz,1h),7.23-7.25(m,1h),7.29(td,j=8.0,1.5hz,2h),7.40-7.45(m,7h),7.48-7.51(m,2h),7.97(s,1h),8.04(t,j=8.5hz,2h),8.09(s,1h),8.11(s,1h),8.13(s,1h),8.22(s,1h)。
配体1的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入化合物4(201mg,0.41mmol,1.0当量),化合物5(126mg,0.45mmol,1.1当量),三(二亚苄基丙酮)二钯(15mg,0.016mmol,4mol%),配体johnphos(10mg,0.032mmol,8mol%),叔丁醇钠(79mg,0.82mmol,2.0当量),然后抽换氮气三次,在氮气保护下加入甲苯(20ml)。该混合物在100℃油浴中搅拌反应2天,薄层色谱监测至原料反应完毕,冷却至室温。过滤,并用乙酸乙酯淋洗,滤液用水洗涤两次,水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥。减压蒸馏除去溶剂,所得粗品用硅胶层析柱分离提纯,洗脱剂:石油醚/二氯甲烷=10:1-1:1,得到配体1,泡沫状固体234mg,收率78%。hrms(esi):c50h33n6o[m+h]+的计算值为733.2710,实测值为733.2715。1hnmr(500mhz,cdcl3):δ7.10-7.03(m,1h),7.11(dd,j=8.5,2.0hz,1h),7.28(td,j=7.0,1.0hz,2h),7.31-7.45(m,13h),7.49(t,j=2.5hz,1h),7.59(dd,j=5.5,2.0hz,1h),7.63(d,j=8.0hz,2h),7.78(d,j=2.5hz,1h),7.89-7.90(m,2h),7.94(s,1h),8.09-8.10(m,2h),8.11-8.12(m,2h),8.14(s,1h),8.16(d,j=0.5hz,1h),8.89(d,j=5.5hz,1h).13cnmr(125mhz,cdcl3):δ103.49,108.96,109.58,109.79,111.14,113.02,113.88,115.03,116.34,118.21,120.01,120.09,120.27,120.61,121.19,121.31,121.36,121.61,121.87,123.24,123.64,124.26,124.32,126.01,126.09,126.49,130.61,138.46,139.33,139.79,140.48,141.14,141.19,147.89,151.20,153.47,155.12,159.44。
pt1的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入配体1(50mg,0.068mmol,1.0当量),亚氯铂酸钾(31mg,0.075mmol,1.1当量),四丁基溴化铵(2mg,0.007mmol,10mol%),然后抽换氮气三次,在氮气保护下加入醋酸(10ml),氮气鼓泡25分钟。该混合物在室温下搅拌8小时,再在110℃的油浴锅中搅拌反应2天,冷却至室温。然后减压蒸馏除去溶剂,所得粗品用硅胶层析柱分离提纯,洗脱剂:石油醚/二氯甲烷=10:1-5:1,得到pt1,黄色固体21mg,收率28%。hrms(esi):c50h33n6195pt[m+h]+的计算值为926.2202,实测值为926.2218。1hnmr(500mhz,dmso-d6):δ7.08(d,j=8.0hz,1h),7.28-7.32(m,2h),7.34-7.39(m,6h),7.47-7.54(m,4h),7.65-7.69(m,3h),7.71(dd,j=6.5,2.5hz,1h),7.84(d,j=8.0hz,2h),7.94(d,j=8.5hz,1h),8.16-8.19(m,2h),8.26-8.30(m,4h),8.40(d,j=2.0hz,1h),8.87(s,1h),9.54(d,j=6.0hz,1h),9.60(s,1h)。13cnmr(125mhz,cdcl3):δ106.16,109.53,109.87,111.78,113.62,114.24,114.94,115.77,115.98,116.72,120.14,120.54,120.69,120.80,121.07,121.79,122.24,123.36,123.51,124.23,124.26,124.61,124.88,126.46,126.73,129.21,135.54,138.28,138.92,141.22,142.28,146.06,147.34,150.20,152.76,153.24,153.67。
合成实施例2:四齿环金属钯(ii)配合物磷光发光材料pd1合成路线
pd1的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入配体1(73mg,0.10mmol,1.0当量),醋酸钯(25mg,0.11mmol,1.1当量),四丁基溴化铵(3mg,0.01mmol,10mol%),然后抽换氮气三次,在氮气保护下加入醋酸(10ml),氮气鼓泡25分钟。该混合物在室温下搅拌8小时,再在110℃的油浴锅中搅拌反应2天,冷却至室温。然后减压蒸馏除去溶剂,所得粗品用硅胶层析柱分离提纯,洗脱剂:石油醚/二氯甲烷=10:1-5:1,得到pd1,白色固体46mg,收率55%。hrms(esi):c50h31n6o106pd[m+h]+的计算值为837.1589,实测值为837.1599。1hnmr(500mhz,dmso-d6):δ7.12(d,j=8.0hz,1h),7.29-7.36(m,8h),7.46-7.53(m,4h),7.64-7.67(m,3h),7.74(d,j=4.5hz,1h),7.81(d,j=8.5hz,2h),7.99(d,j=8.5hz,1h),8.16(d,j=7.5hz,2h),8.25-8.31(m,5h),8.84(s,1h),9.35(d,j=6.0hz,1h),9.58(s,1h)。13cnmr(125mhz,cdcl3):δ106.16,109.52,109.81,110.17,111.43,113.44,114.08,114.59,115.38,116.89,117.50,119.99,120.50,120.57,120.62,121.03,121.66,122.70,123.13,123.34,123.43,124.30,124.48,125.96,126.38,126.63,129.05,135.50,138.25,138.95,141.20,143.36,146.46,147.94,151.03,151.74,152.41,153.25。
光物理测试和理论计算说明:
吸收光谱在agilent8453紫外-可见光光谱仪上测量,使用horibajobinyvonfluorolog-3光谱仪上进行稳态发射实验和寿命测量。低温(77k)发射光谱和寿命在用液氮冷却的2-甲基四氢呋喃溶液中测量。pd(ii)配合物使用gaussian09软件包进行理论计算,利用密度泛函理论(dft)优化了基态(s0)分子的几何结构,使用b3lyp泛函进行dft计算,其中c、h、o和n原子使用6-31g(d)基组,pd原子使用lanl2dz基组。光稳定性测试条件为5%的发光材料:聚苯乙烯薄膜在375nm紫外光激发下(光强:500w/m2)的发光强度衰减。
理论数据及实验数据分析:
表1:基于b3lyp方法优化s0和t1分子构型的pt(ii)和pd(ii)配合物的二面角(°)及其能级
表2:四齿环金属pt(ii)和pd(ii)配合物及对照物的最低激发单线态和三线态的能级差δest
表3:四齿环金属pt(ii)和pd(ii)配合物发光材料光物理性质数据列表
注:λ为发射波长;τobs为材料激发态寿命;φpl为磷光量子效率;kr为辐射速率;其中kr=φpl/τobs。
通过密度泛函理论(dft)计算得到的pt1和pd1的优化分子结构(图1)后的pt1和pd1在其基态(s0)和激发态(t1)下的各芳环之间的二面角变化不大(表1),说明此类分子受激发后分子的几何结构改变较小,利于减小非辐射跃迁。同时又表二可知,与未带给体单元的对照物分子相比,具有给体-受体结构pt1和pd1具有明显减小的最低单线态激发态(s1)和最低单线态激发态(t1)之间的能级δest,利于提高分子的系间窜越速率,进而提高辐射速率kr和缩短激发态寿命τobs;同时提高材料分子的磷光量子效率。
由表3中的光物理数据可知,在二氯甲烷溶液中,与对照物铂配合物(kr=16.3×104s-1)相比,基于给体-受体结构的pt1辐射速率可提高至23.5×104s-1,为原来的1.44倍;在pmma薄膜中,给体-受体结构的pt1辐射速率可提高约4倍。对于钯配合物pd1,相比未带给体的对照物pd可分别提高至原来的8.5倍和10倍。
由图2中pt1和pd1与其配体的吸收光谱比较可以明显看到二者在250-450nm之间增强的金属到配体的电荷转移跃迁吸收(mlct),说明二者具有高效的系间窜越。
由图3和图4中pt1和pd1的低温和室温发射光谱比较可以明显看到二者的延迟荧光发光(df)成分。
由图5中pt1的光稳定性测试可知,其在175分钟时,仍可保持约82%的起始亮度,说明其具有很高的光稳定性。测试条件:5%的发光材料:聚苯乙烯薄膜在375nm紫外光激发下(光强:500w/m2)的发光强度衰减。
以上实验数据和理论计算结果充分表明,本发明申请的给体-受体机构设计可以显著改进四齿环金属铂(ii)或钯(ii)配合物发光材料的光物理性质,缩短激发态寿命短、提高辐射跃迁速率大和磷光量子效率高,使其在oled领域有着巨大的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!