聚氧乙烯聚氧丙烯胺醚、羧酸类聚合物及制备方法和应用与流程
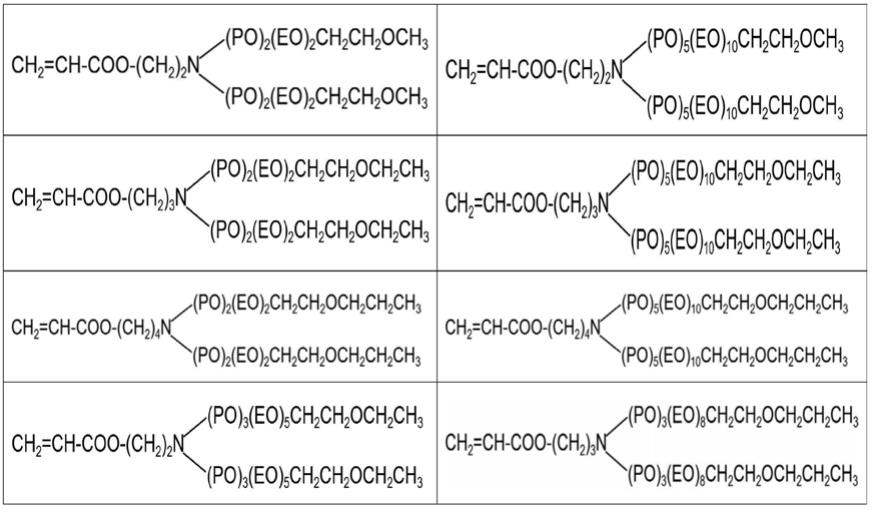
1.本发明属于功能高分子材料制备技术领域,具体涉及一种聚氧乙烯聚氧丙烯胺醚、羧酸类聚合物及制备方法和应用。
背景技术:
2.农药作为防治病虫草害、保护作物的重要手段,在农业生产中起着不可替代的重要作用,但随着人们环保意识与安全意识的不断增强,农药公害问题成为人们日益关注的环境问题。农药制剂领域的研究热点是通过农药制剂技术的创新以达到降低农药对环境的污染。农药制剂开发正向着安全性化,绿色环保化和价格低廉的水基性制剂与无粉尘的固体颗粒制剂方向发展。其中水悬浮剂(sc)或水分散粒剂(wdg)是近几年发展最快,加工工艺最为成熟,成本最低,可加工的农药活性成分最多的剂型。其是在分散剂的作用下,将不溶于水或难溶于水的原药分散到水中形成均匀稳定的分散体系,该剂型与水任意比例均匀混合分散,几乎不受水质和水温的影响,从而能够避免了粉尘污染、危害操作者及对周围环境安全,被认为是二十一世纪最具发展前景的农药剂型之一。
3.聚羧酸盐分散剂是一种新型高效农用分散剂,广泛用于水悬浮剂或水分散粒剂中,该分散剂具有长碳链和较多的吸附位点,支链基团如羧基、磺酸基、氨基以及聚氧乙烯基侧链等可以起到空间排斥作用,因此它的特殊结构对悬浮体系有很好的分散性能。传统的聚羧酸分散剂一般是单臂且含醚键或酯键侧链结构,合成聚羧酸分散剂的原料主要有聚乙二醇、烯丙基聚乙二醇醚等,然而其对于农药制剂的稳定性和悬浮性提升效果一般,为了更好地提高农药制剂的稳定性和悬浮性,有必要开发新的原料,用于制备新的聚羧酸盐分散剂。
技术实现要素:
4.因此,本发明要解决的技术问题在于克服现有技术中的采用传统的聚羧酸盐分散剂导致农药制剂具有稳定性较低、悬浮性较差的缺陷,从而提供一种聚氧乙烯聚氧丙烯胺醚、羧酸类聚合物及制备方法和应用。
5.为此,本发明提供如下技术方案:
6.本发明提供了一种聚氧乙烯聚氧丙烯胺醚,具有式(iii)所示的结构:
[0007][0008]
其中,p为2
‑
4之间的整数,q为0
‑
2之间的整数,m为2
‑
5之间的整数,n为2
‑
10之间的整数。
[0009]
(po)
m
(eo)
n
是指整个(po)
m
(eo)
n
中有m个丙氧基单元po和n个乙氧基单元eo,其中
po和eo随机分布或者统计学分布。
[0010]
可以通过本领域技术人员已知的工艺,例如通过加料顺序调节或同时加入环氧乙烷和环氧丙烷,并控制彼此的摩尔比,得到所需分布的(po)
m
(eo)
n
片段。
[0011]
在优选的实施方式中,所述聚氧乙烯聚氧丙烯胺醚具有如下所示的结构:
[0012][0013][0014]
本发明还提供了一种聚氧乙烯聚氧丙烯胺醚的制备方法,包括如下步骤:
[0015]
(1)将乙二醇烷基醚、环氧乙烷和环氧丙烷在碱性催化剂的作用下反应,得到中间体1;
[0016]
(2)取中间体1与氯化亚砜在碱性催化剂的作用下反应,制得中间体2;
[0017]
(3)取中间体2、碘化钾以及烷醇胺在碱性催化剂的作用下经第一次反应,得到反应液,取反应液与丙烯酰卤经第二次反应,制得聚氧乙烯聚氧丙烯胺醚。
[0018]
其中中间体1具有式(i)的结构通式:
[0019]
h(po)
m
(eo)
n
ch2ch2o(ch2)
q
ch3[0020]
式(i);
[0021]
其中,q为0
‑
2之间的整数,m为2
‑
5之间的整数,n为2
‑
10之间的整数。
[0022]
中间体2具有式(ii)的结构通式:
[0023]
cl(po)
m
(eo)
n
ch2ch2o(ch2)
q
ch3[0024]
式(ii);
[0025]
其中,q为0
‑
2之间的整数,m为2
‑
5之间的整数,n为2
‑
10之间的整数。
[0026]
作为优选的实施方式,满足如下a
‑
f中的任意一项或者多项;
[0027]
a、步骤(1)中乙二醇烷基醚、环氧乙烷和环氧丙烷的摩尔比为1:2
‑
10:2
‑
5,步骤(1)的反应温度为100
‑
110℃,反应时间为3
‑
6h;
[0028]
b、步骤(1)中乙二醇烷基醚与步骤(2)中碱性催化剂、氯化亚砜的摩尔比为1:2
‑
4:2
‑
6,步骤(2)的反应温度为30
‑
50℃,反应时间为16
‑
24h。
[0029]
c、步骤(1)中乙二醇烷基醚与步骤(3)中碱性催化剂、碘化钾、烷醇胺、丙烯酰卤的
摩尔比为5:4
‑
10:30
‑
40:2
‑
6:4
‑
12;步骤(3)中,第一次反应是在氮气回流下进行,反应温度为60
‑
80℃,反应时间为16
‑
24h;第二次反应是将丙烯酰卤滴加到0
‑
10℃的反应液中,反应时间为3
‑
5h;
[0030]
d、步骤(1)中,采用的乙二醇烷基醚为乙二醇甲醚、乙二醇乙醚、乙二醇丙醚中的至少一种;
[0031]
e、步骤(2)和/或步骤(3)中,采用的碱性催化剂为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠中的至少一种;
[0032]
f、步骤(3)中,采用的丙烯酰卤为丙烯酰氯、丙烯酰溴、丙烯酰碘中的一种;所述烷醇胺选自乙醇胺、丙醇胺、丁醇胺中的一种
[0033]
本发明还提供了一种羧酸类共聚物或其盐,具有如下式(iv)所示的结构式:
[0034][0035][0036]
其中,a,b,c均为正整数,p为2
‑
4之间的整数,q为0
‑
2之间的整数,m为2
‑
5之间的整数,n为2
‑
10之间的整数;
[0037]
r1,r2独立地选自h,甲基,羧基;
[0038]
r3,r4,r5独立地选自h,甲基,苯基,己基,
‑
ch2c(ch3)3,
‑
conh2,hoch2ch2nhco
‑
,hoch2ch2ch2nhco
‑
,(ch3)2nch2ch2coo
‑
,
‑
conhc(ch3)2ch2so3h,
‑
cooch2ch2n(ch3)2,
‑
ch2so3na,或者,r4,r5形成环,r4,r5分别与基团的亚甲基连接。
[0039]
作为优选的实施方式,所述局羧酸类共聚物具有如下所示结构:
[0040][0041]
本发明还提供了一种所述羧酸类共聚物,其至少包括三种单体,分别为:
[0042]
(1)至少一种聚氧乙烯聚氧丙烯胺醚;
[0043]
(2)至少一种不饱和羧酸和/或其衍生物;
[0044]
(3)至少一种亲油性不饱和共聚单体;
[0045]
所述聚氧乙烯聚氧丙烯胺醚为本发明任一所述的聚氧乙烯聚氧丙烯胺醚或者任一所述的制备方法制得的聚氧乙烯聚氧丙烯胺醚。
[0046]
三种单体片段随机分布或者统计学分布。
[0047]
可以通过本领域技术人员已知的工艺,例如通过加料顺序调节或同时加入三种单体,并控制彼此的摩尔比,得到所需分布。
[0048]
亲油性不饱和共聚单体是含有碳碳不饱和双键且更倾向于在油溶性溶剂中溶解的共聚单体。
[0049]
作为优选的实施方式,所述羧酸类聚合物的数均分子量为5000
‑
50000。
[0050]
本发明还提供了一种聚羧酸类共聚物或其盐的制备方法,包括取本发明任一所述的聚氧乙烯聚氧丙烯胺醚或者任一所述的制备方法制得的聚氧乙烯聚氧丙烯胺醚、不饱和羧酸和/或其衍生物和亲油性不饱和共聚单体,在引发剂、链转移剂以及溶剂体系下发生共聚反应制得。
[0051]
作为优选的实施方式,所述制备方法还满足如下(1)
‑
(3)中的任意一项或者多项:
[0052]
(1)所述的聚氧乙烯聚氧丙烯胺醚、不饱和羧酸和/或其衍生物、亲油性不饱和共聚单体、引发剂和链转移剂的质量比为100:50
‑
300:50
‑
150:5
‑
20:1
‑
15,共聚反应中,反应温度为50
‑
90℃,反应时间为3.5
‑
7h;
[0053]
(2)所述溶剂体系包括水和有机溶剂,优选地,所述有机溶剂为乙腈、丙酮、丁酮、四氢呋喃、乙醇、甲醇、甲酸异丙酯、乙酸甲酯、醋酸乙酯、乙酸异丙酯中的一种或几种的混合溶剂,有机溶剂和水的质量比为1:1
‑
1:5;
[0054]
(3)所述不饱和羧酸为丙烯酸、甲基丙烯酸、顺丁烯二酸和富马酸中的一种或几种的混合物;不饱和羧酸衍生物为顺丁烯二酸酐;和/或,所述的亲油性不饱和共聚单体为1
‑
辛烯、双环戊二烯、苯乙烯、α
‑
甲基苯乙烯、二异丁烯、羟乙基丙烯酰胺、羟丙基丙烯酰胺、丙烯酰胺、乙烯基吡咯烷酮、苯乙烯磺酸钠、2
‑
丙烯酰胺
‑2‑
甲基丙磺酸、甲基丙烯酸二甲基氨基乙酯和烯丙基磺酸钠中的一种或几种的混合物;和/或,所述链转移剂为巯基乙醇、异丙醇、亚硫酸氢钠和甲基丙烯磺酸钠中的一种或几种的混合物;和/或,所述引发剂选自过硫酸盐引发剂、偶氮类引发剂和过氧化物引发剂中的一种或几种的混合物。
[0055]
在具体的实施方式中,过硫酸盐引发剂为过硫酸钾和/或过硫酸铵;过氧化物引发剂为过氧化二苯甲酰、过氧化二乙酰、过氧化二辛酰和过氧化二月桂酰中的一种或几种的混合物;偶氮类引发剂为偶氮二异丁腈、偶氮二异庚腈、偶氮二异丁酸二甲酯、偶氮异丁氰基甲酰胺和偶氮二环己基甲腈中的一种或几种的混合物。
[0056]
本发明还提供了一种聚羧酸类共聚物或其盐在提高物料的分散性或者在作为分散剂中的应用,所述局羧酸类共聚物为本发明中任一所述的聚羧酸类共聚物或所述的制备方法制得的聚羧酸类共聚物或其盐。
[0057]
其中提高物料的分散性,本发明的分散剂可用于一种固体物质向另一种固体物质分散,也可用于向另一种液体物质或者半固体物质分散。例如应用于农药制剂中。
[0058]
本发明还提供了一种农药制剂,包括本发明任一项所述的聚羧酸类共聚物或者所述的制备方法制得的聚羧酸类共聚物。
[0059]
具体的,所述农药制剂为本领域常规的农药制剂,可以但不局限于水悬浮剂或水分散粒剂。
[0060]
本发明技术方案,具有如下优点:
[0061]
1.本发明提供的聚氧乙烯聚氧丙烯胺醚,具有双臂的不饱和烯酯且带有胺醚的结构,作为原料合成对农药具有优异分散性和稳定性的羧酸类聚合物。
[0062]
2.本发明提供的聚氧乙烯聚氧丙烯胺醚的制备方法,先通过乙二醇烷基醚与环氧乙烷和环氧丙烷反应制得羟基封端的聚氧乙烯聚氧丙烯醚,然后通过与氯化亚砜反应制得氯原子封端的聚氧乙烯聚氧丙烯醚,再通过先后与烷醇胺和丙烯酰卤反应,最终制得聚氧乙烯聚氧丙烯胺醚,反应操作方便,高效,且副反应少,反应收率高,产物纯度高。
[0063]
3.本发明提供的聚氧乙烯聚氧丙烯胺醚的制备方法,步骤(1)中乙二醇烷基醚与步骤(2)中碱性催化剂与氯化亚砜的摩尔比为1:2
‑
4:2
‑
6,并控制反应温度为30
‑
50℃,反应时间为16
‑
24h,通过上述各物质摩尔比联合反应条件的控制,能够明显提高氯代基的取代率,从而提高中间体2的收率和纯度,进而促进聚氧乙烯聚氧丙烯胺醚的生成。
[0064]
4.本发明提供的聚氧乙烯聚氧丙烯胺醚的制备方法,步骤(3)中,第一次反应是在氮气回流下进行,反应温度为60
‑
80℃,反应时间为16
‑
24h;第二次反应是将丙烯酰氯滴加到0
‑
10℃的反应液中,反应时间为3
‑
5h;步骤(1)中乙二醇烷基醚与步骤(3)中碱性催化剂、碘化钾、烷醇胺、丙烯酰卤的摩尔比为5:4
‑
10:30
‑
40:2
‑
6:4
‑
12,通过上述各物质摩尔比以及反应时间和温度的控制,能够促进中间体2中伯氨基上的两个氢原子被完全取代,从而促进双臂聚氧乙烯聚氧丙烯胺醚的合成,提高目标产物的收率和纯度。
[0065]
5.本发明提供的聚氧乙烯聚氧丙烯胺醚的制备方法,步骤(1)中,乙二醇烷基醚、环氧乙烷和环氧丙烷的摩尔比为1:2
‑
10:2
‑
5,环氧乙烷和环氧丙烷的引入可以抑制农药粒子的奥氏熟化过程,提高稳定性,且研究发现可以通过调整环氧乙烷和环氧丙烷到上述比例可以更适应于实现不同种类的农药粒子。
[0066]
6.本发明提供的羧酸类聚合物,不仅具有优异的分散性、崩解性和稳定性,而且颜色为无色或者淡黄色,可以满足对色泽要求度高的场合使用。
[0067]
7.本发明提供的聚氧乙烯聚氧丙烯胺醚的制备方法,发现在水和有机溶剂的混合体系下进行反应,可以有效避免爆聚或者分层的现象,反应易于控制和操作,有效避免了单独采用水或者有机溶剂存在的反应不均匀、分层、溶剂用量过大带来安全风险等问题。
[0068]
8.本发明提供的聚氧乙烯聚氧丙烯胺醚的制备方法,所述的聚氧乙烯聚氧丙烯胺醚、不饱和羧酸和/或其衍生物、亲油性不饱和共聚单体、引发剂和链转移剂的质量比为100:50
‑
300:50
‑
150:5
‑
20:1
‑
15以及有机溶剂和水的质量比为1:1
‑
1:5,共聚反应中,反应温度为50
‑
90℃,反应时间为3.5
‑
7h;通过上述各单体质量以及引发剂和链转移剂质量的控制,溶剂中有机溶剂与水的质量比,以及反应温度和时间,获得了合适分子量的羧酸类聚合物,其数均分子量为5000
‑
50000,该分子量的羧酸类聚合物,能够在最大程度上发挥分散和稳定农药粒子的效果,若分子量过低,其分子侧链所产生的空间位阻太小,农药粒子易发生团聚;若分子量过高,其过长的分子侧链相互之间易发生链缠结,从而降低制剂的稳定性。
具体实施方式
[0069]
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
[0070]
实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
[0071]
本发明中氯化封端聚氧乙烯聚氧丙烯醚中间体2和不饱和双键的聚氧乙烯聚氧丙烯胺醚中间体3的纯度均采用高效液相色谱仪(hplc)测定,测试条件如下:岛津高效液相色谱仪lc
‑
20a系统,流动相(体积比)甲醇/水溶液(75/25),流速1.0ml/min,色谱柱inertsil ods
‑
sp,5μm,4.6mm
×
150mm,进样体积25μl,柱温40℃,检测波长360nm,等度洗脱。
[0072]
实施例1
[0073]
本实施例提供一种聚氧乙烯聚氧丙烯胺醚,其结构式如下式所示:
[0074][0075]
其制备方法包括如下步骤:
[0076]
(1)将乙二醇甲醚76kg(1kmol)和碱性催化剂koh 5.6kg混合,升温至100℃,加入环氧乙烷88kg(2kmol)和环氧丙烷116kg(2kmol),1h加完料后维持100℃继续反应3h,得到280kg的中间体1,经核磁共振氢谱仪测试中间体1中eo聚合度为2,po聚合度为2,数均分子量为280。
[0077]
(2)取238kg氯化亚砜(2kmol)溶于156kg的二甲基亚砜中,得到氯化亚砜溶液。将280kg中间体1和276kg碳酸钾(2kmol)置于500l的二甲基亚砜中,滴加上述氯化亚砜溶液,
在30℃下反应16h,过滤除盐减压蒸馏后得到320kg氯化封端聚氧乙烯聚氧丙烯醚中间体2,收率为62%,纯度为90%。
[0078]
(3)取320kg氯化封端聚氧乙烯聚氧丙烯醚中间体2(1kmol),溶于328kg乙腈中,然后加入110kg碳酸钾(0.8kmol)、996kg碘化钾(6kmol)以及25kg乙醇胺(0.4kmol),60℃氮气回流下反应,反应16h,过滤后,滴入72kg的质量百分数为50%的丙烯酰氯溶液(溶剂为乙腈),在0℃下反应3h,过滤减压蒸馏后得到383kg聚氧乙烯聚氧丙烯胺醚,收率为25%,纯度为92%。
[0079]
本实施例提供一种羧酸类共聚物,作为聚羧酸盐分散剂,其结构式如下式所示:
[0080][0081]
其制备方法包括如下步骤:
[0082]
在50℃的甲醇与水的混合体系中,甲醇60kg,水60kg。向混合体系中加入引发剂过硫酸钾2kg、链转移剂巯基乙醇0.4kg。取40kg聚氧乙烯聚氧丙烯胺醚、20kg甲基丙烯酸和20kg苯乙烯混合后滴入上述反应体系中,2h滴加完毕,50℃继续保温反应1.5h。经质量百分数为32%的naoh水溶液中和至ph为5.5,在50℃下减压蒸馏除去溶剂,得到数均分子量为49800(gpc测得)的聚羧酸盐分散剂agc
‑
1。
[0083]
实施例2
[0084]
本实施例提供一种聚氧乙烯聚氧丙烯胺醚,其结构式如下式所示:
[0085][0086]
其制备方法包括如下步骤:
[0087]
(1)将乙二醇乙醚90kg(1kmol)和碱性催化剂koh 5.6kg混合,升温至110℃,加入环氧乙烷440kg(10kmol)和环氧丙烷290kg(5kmol),2h加完料后反应4h,得到820kg中间体1,核磁共振氢谱仪测试中间体1中eo聚合度为10、po聚合物为5,数均分子量为800。
[0088]
(2)取476kg氯化亚砜(4kmol)溶于250kg的二氯甲烷中,得到氯化亚砜溶液。将820kg中间体1(1kmol)和552kg碳酸钾(4kmol)置于800l二氯甲烷中,滴加上述氯化亚砜溶液,在50℃下反应24h,过滤除盐减压蒸馏后得到970kg氯化封端聚氧乙烯聚氧丙烯醚中间体2,收率为75%,纯度为89%。
[0089]
(3)取970kg氯化封端聚氧乙烯聚氧丙烯醚中间体2(1kmol),溶于500kg四氢呋喃中,然后加入276kg碳酸钾(2kmol)、1328kg碘化钾(8kmol)以及74kg乙醇胺(1.2kmol),80℃氮气回流下反应,反应24h,过滤后,滴入216kg的质量百分数为60%的丙烯酰氯溶液(溶剂为四氢呋喃),在10℃下反应24h,过滤减压蒸馏后得到1390kg聚氧乙烯聚氧丙烯胺醚,收率为48%,纯度为91%。
[0090]
本实施例提供一种羧酸类共聚物,可作为聚羧酸盐分散剂使用,其结构式如下式所示:
[0091][0092]
其制备方法包括如下步骤:
[0093]
在80℃的丁酮与水的混合体系中,丁酮20kg,水100kg。向混合体系中加入引发剂过氧化二乙酰8kg、链转移剂异丙醇6kg。混合滴入40kg不饱和双键的聚氧乙烯聚氧丙烯胺醚中间体3、60kg丙烯酸、60kg顺丁烯二酸酐和60kg苯乙烯磺酸钠,4h滴加完毕,80℃继续保温反应5h。经三乙醇胺中和至ph为8,在80℃下减压蒸馏除去溶剂,得到数均分子量为5120(gpc测得)的聚羧酸盐分散剂agc
‑
2。
[0094]
实施例3
[0095]
本实施例提供一种聚氧乙烯聚氧丙烯胺醚,其结构式如下式所示:
[0096][0097]
其制备方法包括如下步骤:
[0098]
(1)将乙二醇丙醚104kg(1kmol)和碱性催化剂koh 5.6kg混合,升温至105℃,加入环氧乙烷220kg(5kmol)和环氧丙烷174kg(3kmol),1h加完料后反应4h,得到498kg中间体1,经核磁共振氢谱仪测试中间体1中eo聚合度为5、po聚合物为3,数均分子量为500。
[0099]
(2)取357kg氯化亚砜(3kmol)溶于200kg的丙酮中,得到氯化亚砜溶液。将498kg中间体1(1kmol)和414kg碳酸钾(3kmol)置于600l丙酮中,滴加上述氯化亚砜溶液,在40℃下反应20h,过滤除盐减压蒸馏后得到633kg氯化封端聚氧乙烯聚氧丙烯醚中间体2,收率为74%,纯度为97%。
[0100]
(3)取633kg氯化封端聚氧乙烯聚氧丙烯醚中间体2(1kmol),溶于350kg甲酸异丙酯中,然后加入170kg碳酸钠(1.6kmol)、1162kg碘化钾(7kmol)以及49kg乙醇胺(0.8kmol),70℃氮气回流下反应,反应20h,过滤后,滴入144kg的质量百分数为55%的丙烯酰氯溶液(溶剂为甲酸异丙酯),在5℃下反应20h,过滤减压蒸馏后得到915kg聚氧乙烯聚氧丙烯胺醚,收率为42%,纯度为90%。
[0101]
本实施例提供一种羧酸类共聚物,可作为聚羧酸盐分散剂使用,其结构式如下式所示:
[0102][0103]
其制备方法包括如下步骤:
[0104]
在70℃的乙腈与水的混合体系中,乙腈40kg,水80kg。向混合体系中加入引发剂偶氮二异丁腈4kg、链转移剂甲基丙烯磺酸钠3kg。混合滴入40kg不饱和双键的聚氧乙烯聚氧丙烯胺醚中间体3、30kg顺丁烯二酸、30kg顺丁烯二酸酐、20kg羟乙基丙烯酰胺和20kg乙烯基吡咯烷酮,3h滴加完毕,70℃继续保温反应2h。经质量百分数为32%的氢氧化钾溶液中和至ph为7,在70℃下减压蒸馏除去溶剂,得到数均分子量为36930(gpc测得)的聚羧酸盐分散剂agc
‑
3。
[0105]
实施例4
[0106]
本实施例提供一种聚氧乙烯聚氧丙烯胺醚,其结构式如下式所示:
[0107][0108]
其制备方法基本与实施例1相同,区别仅在于步骤(3),本实施例的步骤(3)为取320kg氯化封端聚氧乙烯聚氧丙烯醚中间体2(1kmol),溶于328kg乙腈中,然后加入110kg碳酸钾(0.8kmol)、996kg碘化钾(6kmol)以及30kg丙醇胺(0.4kmol),60℃氮气回流下反应,反应16h,过滤后,滴入72kg的质量百分数为50%的丙烯酰氯溶液(溶剂为乙腈),在0℃下反应3h,过滤减压蒸馏后得到385kg聚氧乙烯聚氧丙烯胺醚,收率为25%,纯度为91%。
[0109]
本实施例提供一种羧酸类共聚物,可作为聚羧酸盐分散剂使用,其结构式如下式所示:
[0110][0111]
其制备方法包括如下步骤:
[0112]
在50℃的甲醇与水的混合体系中,甲醇60kg,水60kg。向混合体系中加入引发剂过硫酸钾2kg、链转移剂巯基乙醇0.4kg。取40kg不饱和双键的聚氧乙烯聚氧丙烯胺醚中间体3、20kg甲基丙烯酸和20kg苯乙烯混合后滴入上述反应体系中,2h滴加完毕,50℃继续保温反应1.5h。经质量百分数为32%的naoh水溶液中和至ph为5.5,在50℃下减压蒸馏除去溶剂,得到数均分子量为46600(gpc测得)的聚羧酸盐分散剂agc
‑
4。
[0113]
实施例5
[0114]
本实施例提供了一种聚氧乙烯聚氧丙烯胺醚和羧酸类聚合物的制备方法,基本与
实施例1一致,区别仅在于羧酸类聚合物的制备过程中,采用向混合体系中加入引发剂过硫酸钾1kg、链转移剂巯基乙醇0.2kg,其余试剂和反应条件均与实施例1一致,得到数均分子量为63200(gpc测得)的聚羧酸盐分散剂。
[0115]
实施例6
[0116]
本实施例提供了一种聚氧乙烯聚氧丙烯胺醚和羧酸类聚合物的制备方法,基本与实施例1一致,区别仅在于羧酸类聚合物的制备过程中,采用向混合体系中加入引发剂过硫酸钾10kg、链转移剂巯基乙醇8kg,其余试剂和反应条件均与实施例1一致,得到数均分子量为3880(gpc测得)的聚羧酸盐分散剂。
[0117]
实施例7
[0118]
本实施例提供了一种聚氧乙烯聚氧丙烯胺醚和羧酸类聚合物的制备方法,基本与实施例1一致,区别仅在于羧酸类聚合物的制备过程中溶剂体系不同,本实施例的溶剂体系为水和甲醇的混合体系,水的质量为110kg,甲醇的质量为10kg,其余试剂和反应条件均与实施例1一致,经反应得到数均分子量为75800(gpc测得)的羧酸类聚合物。
[0119]
实施例8
[0120]
本实施例提供了一种聚氧乙烯聚氧丙烯胺醚和羧酸类聚合物的制备方法,基本与实施例1一致,区别仅在于羧酸类聚合物的制备过程中溶剂体系不同,本实施例的溶剂体系为水和甲醇的混合体系,水的质量为20kg,甲醇的质量为120kg,其余试剂和反应条件均与实施例1一致,经反应得到数均分子量为34860(gpc测得)的羧酸类聚合物。
[0121]
对比例1
[0122]
本实施例提供了一种聚氧乙烯聚氧丙烯胺醚和羧酸类聚合物的制备方法,基本与实施例1一致,区别仅在于制备聚氧乙烯聚氧丙烯胺醚的步骤(1)中,省略环氧丙烷,加入环氧乙烷的量为176kg,其余试剂和反应条件均与实施例1一致。
[0123]
对比例2
[0124]
本实施例提供了一种聚氧乙烯聚氧丙烯胺醚和羧酸类聚合物的制备方法,基本与实施例1一致,区别仅在于制备聚氧乙烯聚氧丙烯胺醚的步骤(3)中,乙醇胺更换成同摩尔的n
‑
甲基乙醇胺,即n
‑
甲基乙醇胺30kg,得到385kg聚氧乙烯聚氧丙烯胺醚,收率为25%,纯度为91%。
[0125]
实验例1
[0126]
将各实施例和对比例所制备的羧酸类聚合物喷雾干燥后所得的粉末作为农药分散粒剂(wdg)中的分散剂,配制质量百分数为90%莠去津原药,具体配制方法为:莠去津原药90kg,本发明中制备的分散剂5kg,润湿剂(703d佳化化学)1kg,干淀粉1kg,其余硅酸镁铝补足至100kg。按上述比例混合,加入气流粉碎机中进行超细粉碎,粒径达2
‑
5μm,得到细粉。加细粉质量10kg的水捏合0.5h后造粒,送入气流烘干机中干燥,经筛分后可得90%莠去津wdg。
[0127]
分别对90%莠去津wdg的粒径、悬浮率、崩解性、分散性和贮藏稳定性进行测试,具体的测试方法为:
[0128]
粒径测试方法为,将1g的90%莠去津wdg分散在100ml水中,用光学显微镜或者激光粒度仪测定其粒径,本实验例采用激光粒度仪进行测定,本发明实施例得到的水悬浮剂的平均粒径大约为0.2微米到5微米。粒径越小,粒剂分散效果越理想。
[0129]
悬浮率(cipac mt161,mt184)测试方法为,取40ml水置于烧杯中,加入4g(w1)90%莠去津wdg,放置30s后,再搅拌90s。接着将它转移至250ml的量筒中,并用水稀释至250ml,上下颠倒量筒15次,静置30分钟后用水泵抽走上部的225ml悬浮液。底部的25ml全部转移至称量盘上并干燥至恒重(w2),悬浮率的计算如下:悬浮率%=(w1
‑
w2)/w2
×
100%。
[0130]
崩解性测试方法为,在250ml量筒中加入100ml水,然后倒入0.5g 90%莠去津wdg,用目视观察开始检测其润湿性和松散性,直至颗粒沉到量筒底部。按以下指标进行评估。a.几乎所有颗粒都已崩解,优;b.大约半数颗粒已崩解,良;c.很少的颗粒崩解,差。
[0131]
分散性(cipac mt160),目视观察,将1g的莠去津wdg加到装有250ml所需的3倍标准硬水(1026ppm)和温度(30℃)水的250ml量筒中。分散呈云状是比较理想的,分散呈粒状掉落是较差的。
[0132]
贮存稳定性(cipac mt39.3,mt46.3)同其他农药制剂一样,在
‑5±
2℃和54
±
2℃下贮存两周,贮存期间不能发生外观变化和有效成分的降解,并测定其贮存后的的悬浮率,悬浮率>90%才满足要求且悬浮率越高制剂的稳定性越好。
[0133]
检测结果如表1所示。
[0134]
表1不同分散剂在90%莠去津wdg中的性能评价结果
[0135][0136]
注:sd
‑
819为来源于上海是大高分子材料有限公司的聚羧酸类型的分散剂;huntsman 2700为来源于亨斯曼厂家的聚羧酸类型的分散剂。
[0137]
从表中数据可知,根据实施例1
‑
4与实施例5
‑
8对比可知,将羧酸类聚合物分子量以及水/溶剂质量比分别控制在5000
‑
50000以及1:1
‑
5:1时,最终90%莠去津wdg经热储,冷储,常温储藏后的分散性,悬浮率以及崩解性更优;根据实施例1
‑
4与对比例1
‑
2以及市面上
产品对比可知,同时引入适量的po和双侧支链结构,可以使90%莠去津wdg经热储,冷储,常温储藏后分散性,悬浮率以及崩解性均满足需求,且优于市面上同类型产品性能。
[0138]
实验例2
[0139]
将各实施例和对比例所制备的羧酸类聚合物作为农药水悬浮剂中的分散剂,配制质量百分数为50%吡虫啉悬浮剂(sc),具体配制方法为:吡虫啉原药50kg,本发明中制备的分散剂5kg,乳化剂(703d佳化化学)2kg,消泡剂(xep5303佳化化学)0.1kg,黄原胶0.1kg,其余水补足至100kg。取各原料按照上述比例混合,采用砂磨机砂磨并经过滤后可得最终样品,即为50%吡虫啉sc。
[0140]
分别对吡虫啉sc的粒径、悬浮率、分散性、流动性和贮藏稳定性进行测试,具体的测试方法为:
[0141]
粒径测试方法为,将1g的50%吡虫啉sc分散在100ml水中,用光学显微镜或者激光粒度仪测定其粒径,本实验例采用激光粒度仪进行测定,本发明实施例得到的水悬浮剂的平均粒径大约为0.2微米到5微米。粒径越小,悬浮剂越理想。
[0142]
悬浮率(cipac mt161,mt184)测试方法为,取40ml水置于烧杯中,加入4g(w1)50%吡虫啉sc,放置30s后,再搅拌90s。接着将它转移至250ml的量筒中,并用水稀释至250ml,上下颠倒量筒15次,静置30分钟后用水泵抽走上部的225ml悬浮液。底部的25ml全部转移至称量盘上并干燥至恒重(w2),悬浮率的计算如下:悬浮率%=(w1
‑
w2)/w2
×
100%。
[0143]
分散性(cipac mt160),目视观察,将1g的50%吡虫啉sc悬浮剂加到装有250ml所需标准硬水(342ppm)和温度(30℃)水的250ml量筒中。分散呈云状是比较理想的,分散呈粒状掉落是较差的。
[0144]
流动性由制剂的粘度来表征,用b
‑
型
‑
粘度计(ndj
‑
1)测定50%吡虫啉sc悬浮剂的粘度,悬浮剂黏度越低,流动性越好。一般粘度在100
‑
500厘泊之间,具体实施过程中就不存在问题。
[0145]
贮存稳定性(cipac mt39.3,mt46.3)同其他农药制剂一样,在0
±
2℃和54
±
2℃下贮存两周,贮存期间不能发生外观变化和有效成分的降解,并测定其贮存后的的悬浮率,悬浮率>90%才满足要求且悬浮率越高制剂的稳定性越好。
[0146]
检测结果如表2所示。
[0147]
表2不同分散剂在50%吡虫啉sc中的性能评价结果
[0148][0149]
注:sd
‑
811为来源于上海是大高分子材料有限公司的聚羧酸类型的分散剂;agrilan 788为来源于诺力昂厂家的聚羧酸类型的分散剂。
[0150]
从表中数据可知,根据实施例1
‑
4与实施例5
‑
8对比可知,将羧酸类聚合物分子量以及水/溶剂质量比分别控制在5000
‑
50000以及1:1
‑
5:1时,最终50%吡虫啉sc经热储,冷储,常温储藏后的分散性,悬浮率以及流动性更优;根据实施例1
‑
4与对比例1
‑
2以及市面上产品对比可知,同时引入适量的po和双侧支链结构,可以使50%吡虫啉sc经热储,冷储,常温储藏后分散性,悬浮率以及流动性均满足需求,且优于市面上同类型产品性能。
[0151]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1