一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法
【技术领域】
本发明涉及有机合成领域,具体涉及一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法。
背景技术:
苯并噁唑类化合物是一类重要的杂环化合物,其广泛存在于生物活性分子、药物和天然产物中,并具有多种生物和药理特性,如:抗菌、抗炎镇痛、抗结核、抗癌、以及抗艾滋病等。苯并噁唑类化合物在医药和农药合成领域常被用作重要的药效基团,在新型医药制剂和农药开发研究中起着重要的作用。该类化合物具备刚性平面结构,含有共轭π键,近些年在功能材料领域也取得了较大的进展。此外,苯并噁唑类化合物在有机合成中作为催化剂或者特殊配体发挥重要作用。
传统的合成方法主要以邻氨基苯酚为底物,而在邻氨基苯酚制备过程中难以避免苯环的硝化反应,这是一种强烈的放热反应,且需要强腐蚀性的混酸,极易造成温度时失控引发爆炸事故。在苯并噁唑的合成研究发展中,也陆续出现了以邻卤或者邻羟基苯胺化合物的分子内环化以及简单苯并噁唑的结构修饰等途径来构建苯并噁唑类化合物。然而这类方法不仅需要进行初始原料的预官能团化,且大多数反应需要过渡金属催化剂以及配体的参与,其存在金属残留的风险,不利于其在生物医药方面的研究开发。因此,开发一种可替代的较为温和的无金属方法用于苯并噁唑类化合物的制备仍然具有重要的意义。
儿茶酚大多以衍生物的形式存在于自然界中。如茶叶中含有丰富的儿茶素化合物,蔬菜和果实中含有丰富的原儿茶酸化合物,高等双子叶植物中和蕨类植物中含有丰富的绿原酸化合物。以儿茶酚类化合物代替传统的邻氨基苯酚来构建苯并噁唑可以降低工业制备原料时带来的风险。近几年,尽管少量文献已经报道了利用儿茶酚类化合物合成苯并噁唑,但这些合成策略都需要金属催化剂的参与。【参考文献:(a)beegum,s.;panicker,c.y.;
针对上述方法的不足,开发无金属条件下以简单易得的儿茶酚类化合物和胺类化合物为原料合成苯并噁唑类化合物的有效方法,该方法反应时间短、操作简便、底物适用范围广,具有一定的工业应用价值。
技术实现要素:
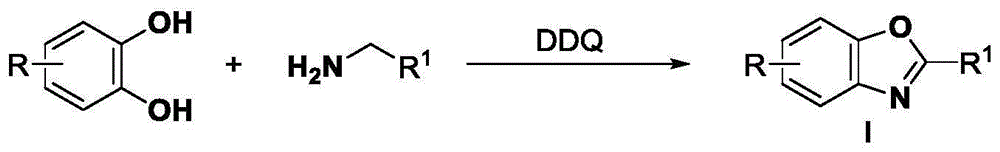
本发明的目的是开发一种在氮气氛围下,以儿茶酚类化合物和胺类化合物为原料,在氧化剂2,3-二氯-5,6-二氰对苯醌(ddq)作用下高转化率地合成苯并噁唑类化合物的方法。
本发明的发明目的是通过如下技术方案实现的:
一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,所述苯并噁唑类化合物的制备原料包括:儿茶酚类化合物、胺类化合物、ddq。
所述苯并噁唑类化合物具有以下结构式(i):
所述结构式中,r为在苯环上的任意一个或多个位置的取代基,且各r独立的选自h、甲基、叔丁基、甲氧基、苯基;r1选自苯基,对甲苯基、对甲氧基苯基、对叔丁基苯基、对氟苯基、对氯苯基、对溴苯基、对碘苯基、对氰基苯基、对三氟甲基苯基、对硝基苯基、苯甲基、叔丁基、甲氧基、1-萘基、2-噻吩基、2-吡啶基、3-吡啶基、4-吡啶基、2-呋喃基、丙基、戊基、异丙基、叔丁基、3-苯基丙基、15烷基、乙烯基、乙炔基、酯基、4-四氢吡喃基、2-氯甲基、2-三氟甲基中的一种。
所述儿茶酚类化合物选自3,5-二叔丁基儿茶酚、对叔丁基儿茶酚、对甲基儿茶酚、对苯基儿茶酚、3-叔丁基-6-甲氧基儿茶酚、儿茶酚中的至少一种。
所述胺类化合物自苄胺、对甲苄胺、对甲氧基苄胺、对叔丁基苄胺、对氟苄胺、对氯苄胺、对溴苄胺、对碘苄胺、对氰基苄胺、对三氟甲基苄胺、对硝基苄胺、苯乙基胺、1-萘甲胺、2-噻吩甲胺、2-吡啶基甲胺、3-吡啶基甲胺、4-吡啶基甲胺、2-呋喃甲胺、正丁胺、正己胺、异丁胺、叔戊胺、3-苯基丁胺、16烷基胺、烯丙胺、炔丙胺、甘氨酸、4-氨甲基四氢吡喃、2-氯乙胺、2,2,2-三氟乙胺中的一种。
优选的,一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,包含以下步骤:取儿茶酚类化合物、胺类化合物、ddq,混合;在惰性气体氛围下,加入溶剂,反应,得到苯并噁唑类化合物。
进一步优选的,一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,包含以下步骤:取儿茶酚类化合物、胺类化合物、ddq,混合;在惰性气体氛围下,加入溶剂,加热搅拌反应,反应结束后冷却至室温,用饱和nacl洗涤,萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得苯并噁唑类化合物。
更优选的,一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,包含以下步骤:取儿茶酚类化合物、胺类化合物、ddq置于反应容器中,混合;在惰性气体氛围下,加入溶剂,加热搅拌反应,反应结束后冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得苯并噁唑类化合物。
所述儿茶酚类化合物、胺类化合物、ddq之间的摩尔比为1:(1.1~1.5):(2~2.3)。
优选的,所述儿茶酚类化合物、胺类化合物、ddq之间的摩尔比为1:(1.1~1.2):(2~2.2)。
更优选的,所述儿茶酚类化合物、胺类化合物、ddq之间的摩尔比为1:1.2:(2~2.1)。
所述溶剂为乙酸乙酯和/或1,2-二氯乙烷。
所述反应的温度为25-70℃。
优选的,所述反应的温度为25-50℃。
所述反应的时间为3-12h。
优选的,所述反应的时间为5-10h。
所述惰性气体为氮气、氩气与氦气中的任意一种或多种的组合。
根据实验研究,本发明提供了一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的方法。该方法具有原料廉价易得、底物范围广、反应体系简单、所得目标产物易分离、反应操作简便、安全可靠等特点。与过渡金属催化的合成方法相比,该体系无需昂贵的金属催化剂和配体,降低了经济成本,避免了重金属对环境造成的污染以及金属在产物中的残留问题。该反应官能团耐受性较高,可用于合成一系列2-芳基、三氟甲基、酯基、卤素、2-烯基、2-炔基、2-烷基取代的苯并噁唑类衍生物,可用于含多种特殊官能团结构的苯并噁唑化合物,具有较大的应用价值。
【附图简要说明】
图1为制备苯并噁唑类化合物的反应式。
【具体实施方式】
下面结合本发明的合成例对本发明所述的合成方法作进一步说明,需要说明的是,实施例并不构成对本发明要求保护范围的限制。
一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,所述苯并噁唑类化合物的制备原料包括:儿茶酚类化合物、胺类化合物、2,3-二氯-5,6-二氰对苯醌(ddq)。
所述苯并噁唑类化合物具有以下结构式(i):
所述结构式中,r为在苯环上的任意一个或多个位置的取代基,且各r独立的选自h、甲基、叔丁基、甲氧基、苯基;r1选自苯基,对甲苯基、对甲氧基苯基、对叔丁基苯基、对氟苯基、对氯苯基、对溴苯基、对碘苯基、对氰基苯基、对三氟甲基苯基、对硝基苯基、苯甲基、叔丁基、甲氧基、1-萘基、2-噻吩基、2-吡啶基、3-吡啶基、4-吡啶基、2-呋喃基、丙基、戊基、异丙基、叔丁基、3-苯基丙基、15烷基、乙烯基、乙炔基、酯基、4-四氢吡喃基、2-氯甲基、2-三氟甲基中的一种。
在一种实施方式中,所述儿茶酚类化合物选自3,5-二叔丁基儿茶酚、对叔丁基儿茶酚、对甲基儿茶酚、对苯基儿茶酚、3-叔丁基-6-甲氧基儿茶酚、儿茶酚中的至少一种。
在一种实施方式中,所述胺类化合物自苄胺、对甲苄胺、对甲氧基苄胺、对叔丁基苄胺、对氟苄胺、对氯苄胺、对溴苄胺、对碘苄胺、对氰基苄胺、对三氟甲基苄胺、对硝基苄胺、苯乙基胺、1-萘甲胺、2-噻吩甲胺、2-吡啶基甲胺、3-吡啶基甲胺、4-吡啶基甲胺、2-呋喃甲胺、正丁胺、正己胺、异丁胺、叔戊胺、3-苯基丁胺、16烷基胺、烯丙胺、炔丙胺、甘氨酸、4-氨甲基四氢吡喃、2-氯乙胺、2,2,2-三氟乙胺中的一种。
在一种实施方式中,一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,包含以下步骤:取儿茶酚类化合物、胺类化合物、ddq,混合;在惰性气体氛围下,加入溶剂,反应,得到苯并噁唑类化合物。
在一种实施方式中,一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,包含以下步骤:取儿茶酚类化合物、胺类化合物、ddq,混合;在惰性气体氛围下,加入溶剂,加热搅拌反应,反应结束后冷却至室温,用饱和nacl洗涤,萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得苯并噁唑类化合物。
在一种实施方式中,一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的合成方法,包含以下步骤:取儿茶酚类化合物、胺类化合物、ddq置于反应容器中,混合;在惰性气体氛围下,加入溶剂,加热搅拌反应,反应结束后冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得苯并噁唑类化合物。
在一种实施方式中,所述儿茶酚类化合物、胺类化合物、ddq之间的摩尔比为1:(1.1~1.5):(2~2.3)。
在一种实施方式中,所述儿茶酚类化合物、胺类化合物、ddq之间的摩尔比为1:(1.1~1.2):(2~2.2)。
在一种实施方式中,所述儿茶酚类化合物、胺类化合物、ddq之间的摩尔比为1:1.2:(2~2.1)。
在一种实施方式中,所述溶剂为乙酸乙酯和/或1,2-二氯乙烷。
在一种实施方式中,所述反应的温度为25-70℃。
在一种实施方式中,所述反应的温度为25-50℃。
在一种实施方式中,所述反应的时间为3-12h。
在一种实施方式中,所述反应的时间为5-10h。
在一种实施方式中,所述惰性气体为氮气、氩气与氦气中的任意一种或多种的组合。
根据实验研究,本发明提供了一种由儿茶酚类化合物和胺类化合物制备苯并噁唑类化合物的方法。该方法具有原料廉价易得、反应体系简单、所得目标产物易分离、反应操作简便、安全可靠等特点。该方法主要解决了苯并噁唑类化合物合成中副产物多、金属残留的难题,具体表现为:以自然界中广泛存在的儿茶酚类生物代替传统的邻氨基苯酚化合物为底物,避免了在制备原料时,硝化过程引发的潜在爆炸危险。该反应底物范围广,官能团耐受性较高,可用于合成一系列2-芳基、2-烯基、2-炔基、2-烷基取代的苯并噁唑类衍生物。同时,三氟甲基、酯基、卤素这些官能团取代的苯并噁唑也能在该体系中获得。本反应的实现可能通过以下途径实现:
下面是具体的合成例。
合成例1
如图1所示,
5,7-二叔丁基-2-苯基苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol苄胺、0.4mmolddq,2.0mlea。在氮气氛围下,在28℃条件下持续搅拌24h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率89%。1hnmr(400mhz,cdcl3):δ8.28-8.25(m,2h),7.67(s,1h),7.54-7.51(m,3h),7.32(s,1h),1.57(s,9h),1.41(s,9h);13cnmr(101mhz,cdcl3):δ162.5,147.7,146.9,142.3,133.7,131.2,128.9,127.6,127.4,119.5,114.2,35.1,34.5,31.8,30.0.合成例2
5,7-二叔丁基-2-(3-氯-4-氟苯基)-苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol3-氯-4-氟苯甲胺、0.4mmolddq,2.0mlea。在氮气氛围下,在28℃条件下持续搅拌24h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率97%。1hnmr(400mhz,cdcl3):δ8.29(d,j=6.9hz,1h),8.15–8.10(m,1h),7.64(s,1h),7.33(s,1h),7.29(t,j=8.6hz,1h),1.54(s,9h),1.39(s,9h);13cnmr(101mhz,cdcl3):δ161.1,159.4(d,j=175.3hz),148.1,147.0,142.1,133.8,129.7,127.3(d,jf-c=7.9hz),124.9(d,jf-c=4.0hz),122.0(d,jf-c=18.4hz),120.0,117.2(d,jf-c=21.8hz),114.3,35.1,34.5,31.7,30.0.
合成例3
5,7-二叔丁基-2-(4-四氢吡喃基)苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol4-氨甲基四氢吡喃、0.4mmolddq,2.0mlea。在氮气氛围下,在28℃条件下持续搅拌24h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率72%。1hnmr(400mhz,cdcl3):δ7.60(s,1h),7.29(s,1h),4.12–4.08(m,2h),3.65–3.58(m,2h),3.29–3.19(m,1h),2.17–2.00(m,4h),1.50(s,9h),1.39(s,9h).;13cnmr(101mhz,cdcl3):δ167.8,147.3,146.8,141.2,133.5,118.9,113.9,67.1,35.0,34.3,31.8,30.0,29.8.
合成例4
5,7-二叔丁基-2-(2-吡啶基)苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol2-吡啶基甲胺、0.4mmolddq,2.0mlea。在氮气氛围下,在28℃条件下持续搅拌24h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率74%。1hnmr(400mhz,cdcl3):δ8.81(d,j=4.5hz,1h),8.27(d,j=7.9hz,1h),7.84(t,j=7.7hz,1h),7.68(s,1h),7.42–7.36(m,1h),7.35(s,1h),1.55(s,9h),1.39(s,9h);13cnmr(101mhz,cdcl3):δ161.0,150.3,147.9,147.2,146.4,142.1,136.7,134.1,125.0,123.0,120.3,114.7,35.0,34.4,31.7,30.0.
合成例5
5,7-二叔丁基-2-氯甲基苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol2-氯乙胺盐酸盐、0.24mmol碳酸氢钠、0.4mmolddq,2.0mlea。在氮气氛围下,在28℃条件下持续搅拌24h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率69%。1hnmr(400mhz,cdcl3):δ7.59(s,1h),7.33(s,1h),4.76(s,2h),1.49(s,9h),1.37(s,9h);13cnmr(101mhz,cdcl3):δ160.2,148.0,147.5,141.1,134.1,120.2,114.5,36.5,35.0,34.4,31.7,29.8.
合成例6
5,7-二叔丁基-2-乙烯基苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol烯丙胺盐酸盐、0.24mmol碳酸氢钠、0.4mmolddq,2.0mlea。在氮气氛围下,在28℃条件下持续搅拌24h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率73%。1hnmr(400mhz,cdcl3):δ7.59(s,1h),7.30(s,1h),6.80-6.72(m,1h),6.44(d,j=17.6hz,1h),5.80(d,j=11.1hz,1h),1.50(s,9h),1.38(s,9h);13cnmr(100mhz,cdcl3):δ161.5,147.6,146.4,142.0,133.5,124.2,124.1,119.8,114.2,34.9,34.3,31.7,29.9.
合成例7
5,7-二叔丁基-2-三氟甲基苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol2,2,2-三氟乙胺、0.4mmolddq,2.0mlea。在氮气氛围下,在28℃条件下持续搅拌24h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率79%。1hnmr(400mhz,cdcl3):δ7.73(s,1h),7.49(s,1h),1.51(s,9h),1.40(s,9h);13cnmr(101mhz,cdcl3):δ151.2(q,jf-c=43.3hz),149.3,147.1,139.8,135.0,122.2,117.0(q,jf-c=269.0hz),115.6,35.1,34.5,31.6,29.8.
合成例8
5,7-二叔丁基苯并[d]噁唑的合成
在反应器中加入0.2mmol3,5-二叔丁基儿茶酚、0.24mmol甘氨酸、0.4mmolddq,2.0mlea。在氮气氛围下,在50℃条件下持续搅拌5h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率80%。1hnmr(400mhz,cdcl3):δ8.07(s,1h),7.66(s,1h),7.35(s,1h),1.49(s,9h),1.39(s,9h);13cnmr(101mhz,cdcl3):δ151.9,147.6,146.2,140.2,134.0,119.8,114.5,34.9,34.3,31.7,29.8.
合成例9
2,5-二叔丁基苯并[d]噁唑的合成
准确称取0.2mmol对叔丁基邻苯二酚,0.4mmolddq加入到烘干的25mlschlenk管中,封管后,抽填氮气各3次,在氮气氛围下加入0.24mmol新戊胺,2.0mlea。反应混合物在室温下(28℃)下反应5h。反应完成后,冷却至室温,反应混合液在真空下浓缩得到粗产品,再用石油醚/乙酸乙酯为20/1的洗脱剂将粗产品经硅胶色谱柱(300-400目)进行柱层析分离提纯,分离产物经过真空干燥后得到40.1mg分析纯的产物,收率:87%,棕色液体。1hnmr(400mhz,cdcl3):δ7.60(d,j=8.4hz,1h),7.52(s,1h),7.35(d,j=8.3hz,1h),1.48(s,9h),1.37(s,9h).13cnmr(100mhz,cdcl3):δ173.2,150.9,148.4,138.7,121.5,118.7,107.0,35.0,34.1,31.7,28.4.
合成例10
5-甲基-2-(4-甲氧基苯基)苯并[d]噁唑的合成
准确称取0.2mmol对甲基邻苯二酚,0.8mmolddq加入到烘干的25mlschlenk管中,封管后,抽填氮气各3次,在氮气氛围下加入0.24mmol4-甲氧基苯甲胺,2.0mlea。反应混合物在室温下(28℃)下反应5h。反应完成后,冷却至室温,反应混合液在真空下浓缩得到粗产品,再用石油醚/乙酸乙酯为20/1的洗脱剂将粗产品经硅胶色谱柱(300-400目)进行柱层析分离提纯,分离产物经过真空干燥后得到28.6mg分析纯的产物,收率:60%,白色固体。1hnmr(400mhz,cdcl3):δ8.17(d,j=8.4hz,2h),7.59(d,j=8.1hz,1h),7.35(s,1h),7.14(d,j=8.1hz,1h),7.01(d,j=8.5hz,2h),3.88(s,3h),2.49(s,3h).13cnmr(100mhz,cdcl3):δ162.6,162.1,150.9,140.0,134.9,129.1,125.5,119.9,118.9,114.3,110.5,55.4,21.7.
合成例11
5-苯基-2-(4-甲氧基苯基)苯并[d]噁唑的合成
准确称取0.2mmol对苯基邻苯二酚,0.4mmolddq加入到烘干的25mlschlenk管中,封管后,抽填氮气各3次,在氮气氛围下加入0.24mmol4-甲氧基苯甲胺,2.0mlea。反应混合物在室温下(28℃)下反应5h。反应完成后,冷却至室温,反应混合液在真空下浓缩得到粗产品,再用石油醚/乙酸乙酯为20/1的洗脱剂将粗产品经硅胶色谱柱(300-400目)进行柱层析分离提纯,分离产物经过真空干燥后得到49.9mg分析纯的产物,收率:83%,熔程:171.8–173.1℃白色固体。1hnmr(400mhz,cdcl3):δ8.21(d,j=8.8hz,2h),7.77(d,j=8.5hz,2h),7.64(d,j=7.3hz,2h),7.58(dd,j=8.2,1.6hz,1h),7.47(t,j=7.6hz,2h),7.37(t,j=7.3hz,1h),7.04(d,j=8.8hz,2h),3.90(s,3h).13cnmr(100mhz,cdcl3):δ163.6162.3,151.3,141.6,140.9,138.4,129.4,128.9,127.4,127.3,124.0,119.5,119.3,114.3,108.9,55.4.
合成例12
4-叔丁基-7-甲氧基-2-苯基苯并[d]噁唑的合成
准确称取0.2mmol3-叔丁基-6-甲氧基邻苯二酚,0.4mmolddq加入到烘干的25mlschlenk管中,封管后,抽填氮气各3次,在氮气氛围下加入0.24mmol苯甲胺,2.0mlea。反应混合物在室温下(28℃)下反应5h。反应完成后,冷却至室温,反应混合液在真空下浓缩得到粗产品,再用石油醚/乙酸乙酯为40/1的洗脱剂将粗产品经硅胶色谱柱(300-400目)进行柱层析分离提纯,分离产物经过真空干燥后得到51.1mg分析纯的产物,收率:91%,熔程:82.4–84.4℃,白色固体。1hnmr(400mhz,cdcl3):δ8.33–8.30(m,2h),7.79–7.38(m,3h),7.14(d,j=8.4hz,1h),6.73(d,j=8.4hz,1h),4.03(s,3h),1.52(s,9h).13cnmr(101mhz,cdcl3):δ161.4,149.9,149.8,132.0,131.1,128.8,127.5,127.4,127.3,121.9,105.2,56.0,33.8,30.0。
- 还没有人留言评论。精彩留言会获得点赞!