聚酯组合物及其应用的制作方法
1.本发明关于一种聚酯组合物,具体来说,是关于一种以特定含量包括特定组分的聚酯组合物。本发明聚酯组合物可作为热塑性高分子的可塑剂。
背景技术:
2.对于部分热塑性高分子而言,为了增进其可塑性、柔软性、可挠性、强韧性等物理性质,需要添加可塑剂以利于加工。所述热塑性高分子例如是聚氯乙烯及其共聚物。可塑剂的实例包括但不限于酞酸酯类(phthalates)、脂肪酸酯类(aliphatic esters)、羧酸酯类(carboxylic esters)、磷酸酯类(phosphates)、环氧化物类(epoxides)、及聚酯(polyester)。
3.一般而言,聚酯可由二元酸与二元醇合成而得到。然而,由此获得的聚酯可塑剂常因副产物的存在而容易发生析出现象,进而不利地影响后续加工产品的外观、物理性质与机械性质。
技术实现要素:
4.本发明提供一种聚酯组合物,其具备良好的保存性(即,不易有析出现象)与耐热性,可作为热塑性高分子的可塑剂,赋予热塑性高分子良好的拉伸强度、伸长率、100%模数等性质。
5.因此,本发明目的之一在于提供一种聚酯组合物,其包括聚酯及第一组分,其中当聚酯组合物在气相层析(gas chromatography,gc)中进行表征时,第一组分在自36.38分钟至36.98分钟的滞留时间被冲提出,第一组分以其在气相层析图谱的相应滞留时间处的层析峰表示,且以聚酯组合物的所有层析峰的总面积计,表示第一组分的层析峰的面积占1.5%至7.0%。
6.于本发明的部分实施方案中,聚酯组合物进一步包括第二组分,其中当聚酯组合物在gc中进行表征时,第二组分在自37.56分钟至38.29分钟的滞留时间被冲提出,第二组分以其在气相层析图谱的相应滞留时间处的层析峰表示,且以聚酯组合物的所有层析峰的总面积计,表示第二组分的层析峰的面积占3.5%至7.5%。
7.于本发明的部分实施方案中,当聚酯组合物在气相层析-质谱(gas chromatography-mass spectrometry,gc-ms)中进行表征时,第一组分在自28.900分钟至29.000分钟的滞留时间被冲提出,且第一组分的碎片模式(fragmentation pattern)包括在质荷比(mass-to-charge ratio,m/z)为选自以下群组的一个或多个信号:29、41、42、43、55、56、83、84、101、111、114、127、129、141、154、156、183、201、273、及400。
8.于本发明的部分实施方案中,第一组分的碎片模式包括在质荷比(m/z)为41、55、56、83、111、114、127、129、141、及201的信号。
9.于本发明的部分实施方案中,当聚酯组合物在gc-ms中进行表征时,第二组分在自29.101分钟至29.200分钟的滞留时间被冲提出,且第二组分的碎片模式包括在质荷比(m/
koh/g之间。
23.于本发明的部分实施方案中,聚酯组合物进一步包括第三组分,其中当聚酯组合物在gc中进行表征时,第三组分在自36.99分钟至37.55分钟的滞留时间被冲提出,第三组分以其在气相层析图谱的相应滞留时间处的层析峰表示,且以聚酯组合物的所有层析峰的总面积计,表示第三组分的层析峰的面积占8.0%至10.0%。
24.于本发明的部分实施方案中,第三组分为下式(iii)所示化合物:
25.[式(iii)]
[0026][0027]
其中,式(iii)的r3为r4为其中*表示键结位置。
[0028]
于本发明的部分实施方案中,当聚酯组合物在gc-ms中进行表征时,第三组分在自29.001分钟至29.100分钟的滞留时间被冲提出,且第三组分的碎片模式包括在质荷比(m/z)为选自以下群组的一个或多个信号:29、41、55、56、69、83、101、111、114、127、129、141、155、183、201、215、287、319、及414。
[0029]
于本发明的部分实施方案中,第三组分的碎片模式包括在质荷比(m/z)为41、55、56、69、111、127、129、141、201、及215的信号。
[0030]
于本发明的部分实施方案中,在上述gc中,聚酯组合物被装入长度60米、内径530微米且膜厚3微米的sge bp1毛细管柱,并在以下条件下进行表征:固定相(stationary phase)为100%二甲基聚硅氧烷;载气(carrier gas)为流速为12毫升/分钟(ml/min)的氮气;升温条件为阶段式,依序为于50℃下维持5分钟,然后以10℃/分钟的升温速率自50℃上升至300℃,接着于300℃下维持40分钟;入口温度为340℃;该聚酯组合物的样品注入体积为1微升(μl);以及检测器为在340℃下操作的火焰离子化检测器(flame ionization detector)。
[0031]
于本发明的部分实施方案中,在上述gc-ms中,聚酯组合物被装入长度30米、内径250微米且膜厚0.25微米的zb-1毛细管柱,以100%二甲基聚硅氧烷为固定相,以流速为1.8毫升/分钟的氦气为载流气体,在以依序于50℃下维持5分钟、然后以10℃/分钟的升温速率自50℃上升至330℃、接着在330℃下持温27分钟的阶段式升温操作后,在350℃的入口温度下,以如下条件操作:电子能量为70ev(电子伏特);离子源温度(ion source temperature)
为250℃;使用四极柱质量分析器(quadrupole mass filter);接口温度(interface temperature)为340℃;质谱扫描范围为29.0m/z至900m/z;以及溶剂延迟为1.5分钟。
[0032]
本发明的另一目的在于提供一种热塑性高分子材料,其包括热塑性高分子及可塑剂,其中可塑剂包括上述聚酯组合物。
[0033]
于本发明的部分实施方案中,热塑性高分子是氯乙烯的均聚物或共聚物。
[0034]
为使本发明的上述目的、技术特征及优点能更明显易懂,下文以部分具体实施方案进行详细说明。
附图说明
[0035]
图1为本发明聚酯组合物的一个实施方案的gc图谱。
[0036]
图2为图1的部分放大示意图。
[0037]
图3为本发明聚酯组合物的一个实施方案的gc-ms图谱。
[0038]
图4为图3的部分放大示意图。
[0039]
图5为层析峰面积的划分方式示意图。
具体实施方式
[0040]
以下将具体地描述本发明的部分具体实施方案;但,在不背离本发明的精神下,本发明尚可以多种不同形式的方案来实践,不应将本发明保护范围限于所述具体实施方案。
[0041]
除非另有说明,于本说明书及申请专利范围中所使用的“一”、“该”及类似用语应理解为包括单数及复数形式。
[0042]
除非另有说明,于本说明书及申请专利范围中所使用的“第一”、“第二”及类似用语仅用于区隔所描述的组件或成分,本身并无特殊涵义,且非意欲指代先后顺序。
[0043]
除非另有说明,于本说明书及申请专利范围中所载“黏度”指在25℃下测得的黏度。
[0044]
除非另有说明,于本说明书及申请专利范围中,用语“常压”指在1大气压(760托(torr))下。
[0045]
本文中,特定组分在特定滞留时间范围内被冲提出的意思是指,表示该特定组分的层析峰的波峰(峰值)落在该特定滞留时间范围内。也即,若峰值落于该滞留时间范围内,则可认定该层析峰属于该组分对应的层析峰。
[0046]
本文中,在gc图谱中,层析峰的面积的划分方式如下:如图5所示,采用b-v-b(基线-峰谷-基线)方式,对所有层析峰设定同一条基线,将特定层析峰的峰谷下拉直线至基线,可划分出该特定层析峰的待积分的面积。基线指无任何测试样品通过检测器时,所检测到的载体气体的信号。
[0047]
本发明对照于现有技术的功效在于,聚酯组合物具备良好的保存性(即,不易有析出现象)与耐热性。以下就本发明聚酯组合物及其相关应用提供详细说明。
[0048]
1.聚酯组合物
[0049]
本发明聚酯组合物具有特定gc图谱特征,且包括聚酯及第一组分,且可进一步包括其他组分(例如第二组分及/或第三组分)。于本发明的部分实施方案中,聚酯组合物更具有特定gc-ms图谱特征。
[0050]
上述聚酯的实例包括但不限于聚己二酸酯、聚丁二酸酯、聚戊二酸酯、聚庚二酸酯、聚辛二酸酯、聚癸二酸酯、聚十一烷二酸酯、及聚十二烷二酸酯。于本发明的部分实施方案中,聚酯为聚己二酸酯,例如衍生自己二酸与新戊二醇及丙二醇的聚酯。聚酯的分子量并无特殊限制,一般而言,聚酯的数量平均分子量可为1200至4000,例如1250、1300、1350、1400、1450、1500、1550、1600、1650、1700、1750、1800、1850、1900、1950、2000、2050、2100、2150、2200、2250、2300、2350、2400、2450、2500、2550、2600、2650、2700、2750、2800、2850、2900、2950、3000、3050、3100、3150、3200、3250、3300、3350、3400、3450、3500、3550、3600、3650、3700、3750、3800、3850、3900、或3950,或介于由上述任两个数值所构成的范围。
[0051]
1.1.聚酯组合物的图谱分析
[0052]
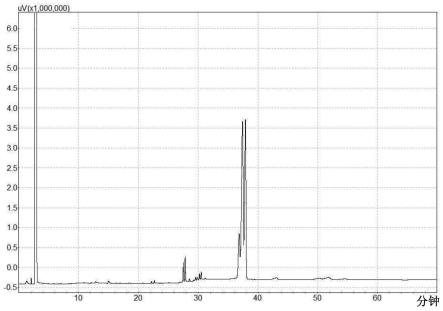
当本发明聚酯组合物在gc中进行表征时,第一组分在自36.38分钟至36.98分钟的滞留时间被冲提出,也即第一组分的层析峰的波峰(峰值)可落在36.38分钟、36.39分钟、36.40分钟、36.41分钟、36.42分钟、36.43分钟、36.44分钟、36.45分钟、36.46分钟、36.47分钟、36.48分钟、36.49分钟、36.50分钟、36.51分钟、36.52分钟、36.53分钟、36.54分钟、36.55分钟、36.56分钟、36.57分钟、36.58分钟、36.59分钟、36.60分钟、36.61分钟、36.62分钟、36.63分钟、36.64分钟、36.65分钟、36.66分钟、36.67分钟、36.68分钟、36.69分钟、36.70分钟、36.71分钟、36.72分钟、36.73分钟、36.74分钟、36.75分钟、36.76分钟、36.77分钟、36.78分钟、36.79分钟、36.80分钟、36.81分钟、36.82分钟、36.83分钟、36.84分钟、36.85分钟、36.86分钟、36.87分钟、36.88分钟、36.89分钟、36.90分钟、36.91分钟、36.92分钟、36.93分钟、36.94分钟、36.95分钟、36.96分钟、36.97分钟、或36.98分钟处,或落在由上述任两个数值所构成的范围内。此外,第一组分以其在gc图谱的相应滞留时间处的层析峰表示,且以聚酯组合物的所有层析峰的总面积计,表示第一组分的层析峰的面积占1.5%至7.0%。图1为本发明聚酯组合物的一实施方案的gc图谱,即,合成例4的聚酯组合物的gc图谱。图2为图1中滞留时间34.5分钟至40.5分钟的部分的放大图。
[0053]
于本发明的部分实施方案中,聚酯组合物进一步包括第二组分,其中当聚酯组合物在gc中进行表征时,第二组分在自37.56分钟至38.29分钟的滞留时间被冲提出,也即第二组分的层析峰的波峰(峰值)可落在37.56分钟、37.57分钟、37.58分钟、37.59分钟、37.60分钟、37.61分钟、37.62分钟、37.63分钟、37.64分钟、37.65分钟、37.66分钟、37.67分钟、37.68分钟、37.69分钟、37.70分钟、37.71分钟、37.72分钟、37.73分钟、37.74分钟、37.75分钟、37.76分钟、37.77分钟、37.78分钟、37.79分钟、37.80分钟、37.81分钟、37.82分钟、37.83分钟、37.84分钟、37.85分钟、37.86分钟、37.87分钟、37.88分钟、37.89分钟、37.90分钟、37.91分钟、37.92分钟、37.93分钟、37.94分钟、37.95分钟、37.96分钟、37.97分钟、37.98分钟、37.99分钟、38.00分钟、38.01分钟、38.02分钟、38.03分钟、38.04分钟、38.05分钟、38.06分钟、38.07分钟、38.08分钟、38.09分钟、38.10分钟、38.11分钟、38.12分钟、38.13分钟、38.14分钟、38.15分钟、38.16分钟、38.17分钟、38.18分钟、38.19分钟、38.20分钟、38.21分钟、38.22分钟、38.23分钟、38.24分钟、38.25分钟、38.26分钟、38.27分钟、38.28分钟、或38.29分钟处,或落在由上述任两个数值所构成的范围内。此外,第二组分以其在气相层析图谱的相应滞留时间处的层析峰表示,且以聚酯组合物的所有层析峰的总面积计,表示第二组分的层析峰的面积占3.5%至7.5%。
[0054]
上述gc分析按照如下方式进行。首先,聚酯组合物被装入长度60米、内径530微米
且膜厚3微米的sge bp1毛细管柱。接着,在以下条件下进行gc分析:固定相为100%二甲基聚硅氧烷;载气为流速为12毫升/分钟的氮气;升温条件为阶段式,依序为于50℃下维持5分钟,然后以10℃/分钟的升温速率自50℃上升至300℃,接着于300℃下维持40分钟;入口温度为340℃;该聚酯组合物的样品注入体积为1微升;以及检测器为在340℃下操作的火焰离子化检测器。
[0055]
在进行gc分析之前,较佳依如下方式将待分析的聚酯组合物样品进行前处理。首先,使聚酯组合物的样品在70℃的烘箱中加热1小时,以使样品均匀。接着,将样品与丙酮(溶剂)混合而配制成重量百分浓度为5%的溶液。
[0056]
于本发明的部分实施方案中,当聚酯组合物在gc-ms中进行表征时,第一组分在自28.900分钟至29.000分钟的滞留时间被冲提出,且第一组分的碎片模式包括在质荷比(m/z)为选自以下群组的一个或多个信号:29、41、42、43、55、56、83、84、101、111、114、127、129、141、154、156、183、201、273、及400,且较佳包括在质荷比为41、55、56、83、111、114、127、129、141、及201的信号。第二组分在自29.1分钟至29.2分钟的滞留时间被冲提出,且第二组分的碎片模式包括在质荷比为选自以下群组的一个或多个信号:29、41、43、55、56、69、83、101、111、127、129、141、147、155、168、197、215、216、343、及428,且较佳包括在质荷比为41、55、56、69、83、111、129、141、155、及215的信号。图3为本发明聚酯组合物的一实施方案的gc-ms图谱,即,合成例4的聚酯组合物的gc-ms图谱。图4为图3中滞留时间26.5分钟至32分钟的部分的放大图。
[0057]
上述gc-ms分析按照如下方式进行。首先,聚酯组合物被装入长度30米、内径250微米且膜厚0.25微米的zb-1毛细管柱。接着,以100%二甲基聚硅氧烷为固定相,以流速为1.8毫升/分钟的氦气为载流气体,在以依序于50℃下维持5分钟、然后以10℃/分钟的升温速率自50℃上升至330℃、接着在330℃下持温27分钟的阶段式升温操作后,在350℃的入口温度下,以如下条件进行gc-ms分析:电子能量为70ev;离子源温度为250℃;使用四极柱质量分析器;接口温度为340℃;质谱扫描范围为29.0m/z至900m/z;以及溶剂延迟为1.5分钟。
[0058]
在进行gc-ms分析之前,较佳依如下方式将待分析的聚酯组合物样品进行前处理。首先,使聚酯组合物的样品在70℃的烘箱中加热1小时,以使样品均匀。接着,将样品与丙酮(溶剂)混合而配制成重量百分浓度为1%的溶液。
[0059]
于本发明的部分实施方案中,聚酯组合物进一步包括第三组分。当聚酯组合物在上述gc分析中进行表征时,第三组分在自36.99分钟至37.55分钟的滞留时间被冲提出,也即第三组分的层析峰的波峰(峰值)可落在36.99分钟、37.00分钟、37.01分钟、37.02分钟、37.03分钟、37.04分钟、37.05分钟、37.06分钟、37.07分钟、37.08分钟、37.09分钟、37.10分钟、37.11分钟、37.12分钟、37.13分钟、37.14分钟、37.15分钟、37.16分钟、37.17分钟、37.18分钟、37.19分钟、37.20分钟、37.21分钟、37.22分钟、37.23分钟、37.24分钟、37.25分钟、37.26分钟、37.27分钟、37.28分钟、37.29分钟、37.30分钟、37.31分钟、37.32分钟、37.33分钟、37.34分钟、37.35分钟、37.36分钟、37.37分钟、37.38分钟、37.39分钟、37.40分钟、37.41分钟、37.42分钟、37.43分钟、37.44分钟、37.45分钟、37.46分钟、37.47分钟、37.48分钟、37.49分钟、37.50分钟、37.51分钟、37.52分钟、37.53分钟、37.54分钟、或37.55分钟处,或落在由上述任两个数值所构成的范围内。此外,以聚酯组合物的所有层析峰的面积计,表示第三组分的层析峰的面积占8.0%至10.0%。此外,当聚酯组合物在上述gc-ms分
析中进行表征时,第三组分在自29.001分钟至29.100分钟的滞留时间被冲提出,且第三组分的碎片模式包括在质荷比为选自以下群组的一个或多个信号:29、41、55、56、69、83、101、111、114、127、129、141、155、183、201、215、287、319、及414,且较佳包括在质荷比为41、55、56、69、111、127、129、141、201、及215的信号。上述gc与gc-ms的分析方式如前文说明,于此不另赘述。
[0060]
1.1.1.第一组分
[0061]
为使本发明聚酯组合物具有良好的保存性及耐热性,以及赋予热塑性高分子良好的拉伸强度、伸长率、100%模数等性质,第一组分在聚酯组合物中的含量被控制在特定范围内。因此,如前文说明,当本发明聚酯组合物在上述gc分析中进行表征时,以聚酯组合物的所有层析峰的总面积计,表示第一组分的层析峰的面积占1.5%至7.0%,例如1.6%、1.7%、1.8%、1.9%、2.0%、2.1%、2.2%、2.3%、2.4%、2.5%、2.6%、2.7%、2.8%、2.9%、3.0%、3.1%、3.2%、3.3%、3.4%、3.5%、3.6%、3.7%、3.8%、3.9%、4.0%、4.1%、4.2%、4.3%、4.4%、4.5%、4.6%、4.7%、4.8%、4.9%、5.0%、5.1%、5.2%、5.3%、5.4%、5.5%、5.6%、5.7%、5.8%、5.9%、6.0%、6.1%、6.2%、6.3%、6.4%、6.5%、6.6%、6.7%、6.8%、或6.9%,或介于由上述任两个数值所构成的范围。相较于其他组分,第一组分的结构的耐热性较差,当第一组分的含量高于前述范围时,会导致热塑性高分子材料的老化后伸长残率不佳。此外,第一组分的结构较柔软,当第一组分的含量低于前述范围时,会导致热塑性高分子材料的伸长率100%模数过高,可塑化效率不佳。
[0062]
于本发明的部分实施方案中,第一组分可为下式(i)所示化合物:
[0063]
[式(i)]
[0064][0065]
于式(i)中,r1为为r1较佳为其中*表示键结位置。式(i)所示化合物可通过使己二酸与2-甲基-1,3-丙二醇、乙二醇、或1,3-丙二醇反应而得到。
[0066]
1.1.2.第二组分
[0067]
为使本发明聚酯组合物具有良好的保存性及耐热性,以及赋予热塑性高分子良好的拉伸强度、伸长率、100%模数等性质,第二组分在聚酯组合物中的含量被控制在特定范
围内。因此,如前文说明,当本发明聚酯组合物在上述gc分析中进行表征时,以聚酯组合物的所有层析峰的总面积计,表示第二组分的层析峰的面积占3.5%至7.5%,例如3.6%、3.7%、3.8%、3.9%、4.0%、4.1%、4.2%、4.3%、4.4%、4.5%、4.6%、4.7%、4.8%、4.9%、5.0%、5.1%、5.2%、5.3%、5.4%、5.5%、5.6%、5.7%、5.8%、5.9%、6.0%、6.1%、6.2%、6.3%、6.4%、6.5%、6.6%、6.7%、6.8%、6.9%、7.0%、7.1%、7.2%、7.3%、或7.4%,或介于由上述任两个数值所构成的范围。第二组分的结构有支链存在而有利于改善聚酯组合物的耐移行性,若第二组分的含量低于前述范围时,聚酯组合物的耐移行性较差。若第二组分的含量高于前述范围时,易导致有结晶析出,聚酯组合物的耐移行性也变差。
[0068]
于本发明的部分实施方案中,第二组分可为下式(ii)所示化合物:
[0069]
[式(ii)]
[0070][0071]
于式(ii)中,r2为r2较佳为其中*表示键结位置。式(ii)所示化合物可通过使己二酸与新戊二醇或1,4-丁二醇反应而得到。
[0072]
1.1.3.第三组分
[0073]
于本发明的部分实施方案中,本发明聚酯组合物进一步包括第三组分。为使本发明聚酯组合物具有良好的保存性及耐热性,以及赋予热塑性高分子良好的拉伸强度、伸长率、100%模数等性质,第三组分在聚酯组合物中的含量较佳被控制在特定范围内。因此,如前文说明,当本发明聚酯组合物在上述gc分析中进行表征时,以聚酯组合物的所有层析峰的总面积计,表示第三组分的层析峰的面积占8.0%至10.0%,例如8.1%、8.2%、8.3%、8.4%、8.5%、8.6%、8.7%、8.8%、8.9%、9.0%、9.1%、9.2%、9.3%、9.4%、9.5%、9.6%、9.7%、9.8%、或9.9%,或介于由上述任两个数值所构成的范围。
[0074]
于本发明的部分实施方案中,第三组分可由下式(iii)所示化合物来表示:
[0075]
[式(iii)]
[0076][0077]
于式(iii)中,r3为r4为其中*表示键结位置。式(iii)所示化合物可通过使己二酸与新戊二醇及2-甲基-1,3-丙二醇反应而得到。
[0078]
1.2.聚酯组合物的制备
[0079]
本发明聚酯组合物通过使一种或多种脂肪族二酸与一种或多种脂肪族二醇进行酯化聚合反应而得到。脂肪族二酸的实例包括但不限于丁二酸、戊二酸、己二酸、庚二酸、辛二酸、癸二酸、十一烷二酸、及十二烷二酸。前述各脂肪族二酸可单独使用,也可任意组合使用。脂肪族二醇的实例包括但不限于乙二醇、1,3-丙二醇、1,4-丁二醇、2-甲基-1,3-丙二醇、新戊二醇、3-甲基-1,5-戊二醇、2,4-二甲基-1,5-戊二醇、2,4-二乙基-1,5-戊二醇、2-乙基-2-丁基-1,3-丙二醇、及2,2,4-三甲基-1,3-戊二醇。前述各脂肪族二醇可单独使用,也可任意组合使用。于本发明的部分实施方案中,使用己二酸与新戊二醇及2-甲基-1,3-丙二醇进行酯化聚合反应。
[0080]
此外,酯化聚合反应通常需要通过添加混合型反应终止剂来终止反应,所述混合型反应终止剂包括但不限于脂肪族单羧酸与脂肪族饱和一元醇的混合物。脂肪族单羧酸的实例包括但不限于正丁酸、异丁酸、正戊酸、异戊酸、正己酸、异己酸、正庚酸、异庚酸、正辛酸、异辛酸、2-乙基己酸、正壬酸、正癸酸、月桂酸、肉豆蔻酸、棕榈酸、及硬脂酸。前述各脂肪族单羧酸可单独使用,也可任意组合使用。脂肪族饱和一元醇的实例包括但不限于正戊醇、异戊醇、正己醇、异己醇、正辛醇、异辛醇、2-乙基己醇、2,2-二甲基戊醇、正壬醇、异壬醇、正癸醇、异癸醇、2,2,4-三甲基戊醇、及月桂醇。前述各脂肪族饱和一元醇可单独使用,也可任意组合使用。于本发明的部分实施方案中,使用月桂酸与2-乙基己醇的混合物。
[0081]
本发明聚酯组合物可在不添加触媒的情况下进行制备。然而,为了缩短反应时间,也可添加触媒。可用于制备本发明聚酯组合物的触媒的实例包括但不限于硫酸、磷酸、对甲苯磺酸、二乙酸二丁基锡、月桂酸二丁基锡、氧化二丁基锡、四异丙基钛酸酯、四丁基钛酸酯、四异辛基钛酸酯、及钛。
[0082]
于本发明的部分实施方案中,是以如下方式制备本发明聚酯组合物。首先,将反应物放入反应器中,在流通氮气并搅拌反应物的条件下,在140℃至200℃进行常压酯化反应且开始脱水。接着,加入触媒,在210℃至230℃及氮气气氛下进行常压酯化反应。之后,将压
力减至460托至200托,在210℃至240℃进行减压酯化反应,当酸价为5mg koh/g以下即表示酯化反应终止。接着,将压力减至0托在210℃至230℃进行减压聚缩合反应,当羟基价介于1.0mg koh/g至15.0mg koh/g之间即表示聚缩合反应终止。最后,降温并进行过滤,得到聚酯组合物。
[0083]
在聚酯组合物中,第一组分、第二组分及第三组分的含量可通过控制:添加原料比例、酯化温度、酯化时间及聚缩合时间等,以获得理想的各组分含量。
[0084]
1.3.聚酯组合物的性质
[0085]
于本发明的部分实施方案中,本发明聚酯组合物具有下述性质,但本发明并不限于此,本发明所属技术领域技术人员可依实际需要调整聚酯组合物的相关性质。聚酯组合物在25℃下的黏度介于2000cps至4000cps之间,例如2050cps、2100cps、2150cps、2200cps、2250cps、2300cps、2350cps、2400cps、2450cps、2500cps、2550cps、2600cps、2650cps、2700cps、2750cps、2800cps、2850cps、2900cps、2950cps、3000cps、3050cps、3100cps、3150cps、3200cps、3250cps、3300cps、3350cps、3400cps、3450cps、3500cps、3550cps、3600cps、3650cps、3700cps、3750cps、3800cps、3850cps、3900cps、或3950cps,或介于由上述任两个数值所构成的范围。聚酯组合物的酸价介于0.1mg koh/g至0.7mg koh/g之间,例如0.15mg koh/g、0.2mg koh/g、0.25mg koh/g、0.3mg koh/g、0.35mg koh/g、0.4mg koh/g、0.45mg koh/g、0.5mg koh/g、0.55mg koh/g、0.6mg koh/g、或0.65mg koh/g,或介于由上述任两个数值所构成的范围。聚酯组合物的羟基价介于5.0mg koh/g至15.0mg koh/g之间,例如5.5mg koh/g、6.0mg koh/g、6.5mg koh/g、7.0mg koh/g、7.5mg koh/g、8.0mg koh/g、8.5mg koh/g、9.0mg koh/g、9.5mg koh/g、10.0mg koh/g、10.5mg koh/g、11.0mg koh/g、11.5mg koh/g、12.0mg koh/g、12.5mg koh/g、13.0mg koh/g、13.5mg koh/g、14.0mg koh/g、或14.5mg koh/g,或介于由上述任两个数值所构成的范围。
[0086]
2.热塑性高分子材料
[0087]
本发明聚酯组合物可作为热塑性高分子的可塑剂,因此,本发明也提供一种热塑性高分子材料,其包括热塑性高分子及可塑剂,所述可塑剂可包括本发明前述聚酯组合物。或者,所述可塑剂可实质上由本发明前述聚酯组合物所构成或由本发明前述聚酯组合物所构成。前述热塑性高分子的实例包括但不限于氯乙烯的均聚物或共聚物,例如氯乙烯-醋酸乙烯酯共聚物、氯乙烯-二氯乙烯共聚物、及氯乙烯-甲基丙烯酸甲酯共聚物。本发明热塑性高分子材料具有良好的拉伸强度、伸长率、100%模数等性质,适合用于各种加工应用。
[0088]
于本发明热塑性高分子材料中,以100重量份的热塑性高分子计,聚酯组合物的含量可为10至120重量份,例如11重量份、12重量份、13重量份、14重量份、15重量份、16重量份、17重量份、18重量份、19重量份、20重量份、21重量份、22重量份、23重量份、24重量份、25重量份、26重量份、27重量份、28重量份、29重量份、30重量份、31重量份、32重量份、33重量份、34重量份、35重量份、36重量份、37重量份、38重量份、39重量份、40重量份、41重量份、42重量份、43重量份、44重量份、45重量份、46重量份、47重量份、48重量份、49重量份、50重量份、51重量份、52重量份、53重量份、54重量份、55重量份、56重量份、57重量份、58重量份、59重量份、60重量份、61重量份、62重量份、63重量份、64重量份、65重量份、66重量份、67重量份、68重量份、69重量份、70重量份、71重量份、72重量份、73重量份、74重量份、75重量份、76重量份、77重量份、78重量份、79重量份、80重量份、81重量份、82重量份、83重量份、84重量
份、85重量份、86重量份、87重量份、88重量份、89重量份、90重量份、91重量份、92重量份、93重量份、94重量份、95重量份、96重量份、97重量份、98重量份、99重量份、100重量份、101重量份、102重量份、103重量份、104重量份、105重量份、106重量份、107重量份、108重量份、109重量份、110重量份、111重量份、112重量份、113重量份、114重量份、115重量份、116重量份、117重量份、118重量份、或119重量份,或介于由上述任两个数值所构成的范围。
[0089]
本发明热塑性高分子材料可视需要包括其他添加剂,以适应性改良热塑性高分子材料的可加工性,或赋予最终产品特定性质。所述添加剂包括但不限于安定剂、填料、染料、颜料、抗氧化剂、阻燃剂、发泡剂、及静电防止剂。
[0090]
于本发明的部分实施方案中,热塑性高分子材料进一步包括安定剂。安定剂的实例包括但不限于环氧化大豆油、钙锌系安定剂、钡锌系安定剂、及铅系安定剂。于本发明热塑性高分子材料中,以100重量份的热塑性高分子计,安定剂的含量可为0重量份至10重量份,例如0.1重量份、0.2重量份、0.3重量份、0.4重量份、0.5重量份、0.6重量份、0.7重量份、0.8重量份、0.9重量份、1重量份、2重量份、3重量份、4重量份、5重量份、6重量份、7重量份、8重量份、或9重量份,或介于由上述任两个数值所构成的范围。
[0091]
3.实施例
[0092]
3.1.测量方式
[0093]
[gc图谱分析]
[0094]
使聚酯组合物的样品在70℃的烘箱中加热1小时,以使样品均匀。接着,将样品与丙酮(溶剂)混合而配制成重量百分浓度为5%的溶液,完成待测样品的前处理。
[0095]
将经前处理过的样品装入长度60米、内径530微米且膜厚3微米的sge bp1毛细管柱,之后将样品放入气相层析质谱仪(型号:gc-2014,购自岛津(shimadzu))中,并在以下条件下操作:固定相为100%二甲基聚硅氧烷;载气为流速为12毫升/分钟的氮气;升温条件为阶段式,依序为于50℃下维持5分钟,然后以10℃/分钟的升温速率自50℃上升至300℃,接着于300℃下维持40分钟;入口温度为340℃;样品注入体积为1微升;以及检测器为在340℃下操作的火焰离子化检测器。
[0096]
[gc-ms图谱分析]
[0097]
使聚酯组合物的样品在70℃的烘箱中加热1小时,以使样品均匀。接着,将样品与丙酮(溶剂)混合而配制成重量百分浓度为1%的溶液,完成待测样品的前处理。
[0098]
将经前处理过的样品装入长度30米、内径250微米且膜厚0.25微米的zb-1毛细管柱,之后将样品放入气相层析质谱仪(型号:qp2020,购自岛津)中,以100%二甲基聚硅氧烷为固定相,以流速为1.8毫升/分钟的氦气为载流气体,在以依序于50℃下维持5分钟、然后以10℃/分钟的升温速率自50℃上升至330℃、接着在330℃下持温27分钟的阶段式升温操作后,在350℃的入口温度下,以如下条件操作:电子能量为70ev;离子源温度为250℃;使用四极柱质量分析器;接口温度为340℃;质谱扫描范围为29.0m/z至900m/z;以及溶剂延迟为1.5分钟。
[0099]
[酸价分析]
[0100]
首先按照以下方式准备koh溶液。称取0.7克(g)的koh并溶于少量蒸馏水中,再以中性乙醇稀释至1000毫升而得到koh溶液。称取0.4克的试药级酞酸氢钾(potassium hydrogen phthalate,khp)并置于250毫升的三角烧瓶中,加入100毫升的蒸馏水使khp溶解
后,滴入2滴酚酞指示剂,以koh溶液滴定,计算得到koh溶液的当量浓度n。
[0101]
接着,称取10克的待测样品并置于250毫升的三角烧瓶中,加入100毫升的中性2-丙醇溶液与3至4滴酚酞指示剂,震荡并稍微加热使样品完全溶解,并以koh溶液进行滴定,直到溶液变成微红色且持续30秒不变色,即为滴定终点。依据以下公式(1)计算样品的酸价。
[0102]
[公式(1)]
[0103][0104]
[羟基价分析]
[0105]
首先依如下方式配制乙酰化试剂。以吸管吸取10毫升的乙酐(acetic anhydride)并置于100毫升的烧杯中,加入50毫升的吡啶,搅拌溶解而得到乙酰化试剂。
[0106]
接着,依以下方式准备koh溶液。称取0.7克的koh并溶于少量蒸馏水中,再以中性乙醇稀释至1000毫升而得到koh溶液。称取0.4克的试药级酞酸氢钾(potassium hydrogen phthalate,khp)并置于250毫升的三角烧瓶中,加入100毫升的蒸馏水使khp溶解后,滴入2滴酚酞指示剂,以koh溶液滴定,记录koh滴定量(即,空白滴定量)并计算得到koh溶液的当量浓度n。
[0107]
之后,称取10克的待测样品并置于共栓三角烧瓶(stoppered conical flask)中,加入10毫升的乙酰化试剂。于烧瓶上放置空气冷却器,在振荡烧瓶的情况下利用水蒸气(温度为98
±
2℃)加热烧瓶1小时。待反应结束后,使烧瓶冷却至室温,加入25毫升的正丁醇并且剧烈摇荡。之后,加入2至3滴的酚酞指示剂,并以koh溶液进行滴定,直到溶液变成微红色且持续30秒不变色,即为滴定终点,记录koh滴定量(即,试验滴定量),并依据以下公式(2)计算样品的羟基价。
[0108]
[公式(2)]
[0109][0110]
[黏度分析]
[0111]
将待测样品倒入高180毫米且直径50毫米的量筒中后,将量筒置于25
±
1℃的恒温水浴中,选择适当的转针与转速,利用布氏lv型黏度计(brookfield viscometer model lv)进行测试。
[0112]
[初始拉伸强度、初始伸长率及初始伸长率100%模数分析]
[0113]
首先,使热压机在170℃下预热5分钟,接着在相同温度下将热塑性高分子材料以50千克/平方厘米(kg/cm2)的压力热压5分钟,以形成长度20厘米、宽度20厘米且厚度1毫米的试片。之后,在60千克/平方厘米的压力下使试片冷却至室温。接着,利用jis-6301-3号型的刀模将试片切成哑铃形试片后,利用万能试验机(型号:3366,购自instron(英斯特朗))根据astm d638的规范对哑铃形试片进行测试,得到初始拉伸强度、初始伸长率与初始伸长率100%模数。
[0114]
[挥发率的分析]
[0115]
首先,使热压机在170℃下预热5分钟,接着在相同温度下将热塑性高分子材料以
50千克/平方厘米(kg/cm2)的压力热压5分钟,以形成长度20厘米、宽度20厘米且厚度1毫米的试片。之后,在60千克/平方厘米的压力下使试片冷却至室温,测量试片的重量。接着,将试片置于136℃的温度下达168小时以进行耐热老化,并测量耐热老化后的试片的重量。依据以下公式(3)计算可得到试片的挥发率(%)。
[0116]
[公式(3)]
[0117][0118]
[耐热老化后拉伸强度、耐热老化后伸长率及耐热老化后伸长率100%模数分析]
[0119]
首先,使热压机在170℃下预热5分钟,接着在相同温度下将热塑性高分子材料以50千克/平方厘米(kg/cm2)的压力热压5分钟,以形成长度20厘米、宽度20厘米且厚度1毫米的试片。接着,利用jis-6301-3号型的刀模将试片切成哑铃形试片,并将哑铃形试片置于136℃的温度下达168小时以进行耐热老化。利用万能试验机3366根据astm d638的规范对耐热老化后的试片进行测试,得到耐热老化后拉伸强度、耐热老化后伸长率及耐热老化后伸长率100%模数。将耐热老化后的拉伸强度除以初始的拉伸强度而得到拉伸强度的残率(本文中也称“拉伸残率(residual tensile rate)”),将耐热老化后的伸长率除以初始的伸长率而得到伸长率的残率(本文中也称“伸长率残率(residual elongation rate)”)。
[0120]
[耐移行性测试]
[0121]
将热塑性高分子材料压制成长度38毫米、宽度6毫米且厚度1毫米的试片,各取两片上下分开置于丙烯腈-丁二烯-苯乙烯树脂板(型号:tairilac ag15e1,购自中国台湾化纤公司)及耐冲击聚苯乙烯树脂板(型号:tairirex hp8250,购自中国台湾化纤公司)两片之间,以三明治试验夹夹住,在其上面荷重1千克的砝码并置于70℃的烘箱达72小时。测试后观察聚酯组合物从热塑性高分子材料所制得的片材是否迁移与浸蚀至树脂板的表面,并以目视评判树脂板表面损伤。判断标准如下:数字“1”表示无痕迹,数字“2”表示有压影,数字“3”表示有明显压痕,数字“4”表示有明显凸痕或凹痕,数字“5”表示表面有严重损伤及有黏性,且数字越低代表耐移行性越佳。
[0122]
3.2.聚酯组合物的制备与性质分析
[0123]
3.2.1.聚酯组合物的制备
[0124]
[合成例1]
[0125]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、240.53克的新戊二醇、485.63克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.22克的四异丙基钛酸酯作为触媒,在220℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在210℃下进行减压酯化反应,酯化反应共历时298分钟,且酯化终点的酸价为4.79mg koh/g。接着,将压力减至0托而在230℃下进行减压聚缩合反应,聚缩合反应共历时397分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土(型号:hyflo)。接着,利用板框式过滤器进行过滤,得到合成例1的聚酯组合物。
[0126]
[合成例2]
[0127]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、320.70克的新戊二醇、416.25克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.22克的四异丙基钛酸酯作为触媒,在220℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在220℃下进行减压酯化反应,酯化反应共历时318分钟,且酯化终点的酸价为4.69mg koh/g。接着,将压力减至0托而在220℃下进行减压聚缩合反应,聚缩合反应共历时465分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土hyflo。接着,利用板框式过滤器进行过滤,得到合成例2的聚酯组合物。
[0128]
[合成例3]
[0129]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、400.88克的新戊二醇、346.88克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.22克的四异丙基钛酸酯作为触媒,在220℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在210℃下进行减压酯化反应,酯化反应共历时310分钟,且酯化终点的酸价为4.87mg koh/g。接着,将压力减至0托而在220℃下进行减压聚缩合反应,聚缩合反应共历时450分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土hyflo。接着,利用板框式过滤器进行过滤,得到合成例3的聚酯组合物。
[0130]
[合成例4]
[0131]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、481.05克的新戊二醇、277.50克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.22克的四异丙基钛酸酯作为触媒,在230℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在220℃下进行减压酯化反应,酯化反应共历时271分钟,且酯化终点的酸价为3.9mg koh/g。接着,将压力减至0托而在210℃下进行减压聚缩合反应,聚缩合反应共历时510分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土hyflo。接着,利用板框式过滤器进行过滤,得到合成例4的聚酯组合物。
[0132]
[合成例5]
[0133]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、481.05克的新戊二醇、277.50克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.22克的四异丙基钛酸酯作为触媒,在220℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在235℃下进行减压酯化反应,酯化反应共历时235分钟,且酯化终点的酸价为2.4mg koh/g。接着,将压力减至0托而在225℃下进行减压聚缩合反应,聚缩合反应共历时413分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土hyflo。接着,利用板框式过滤器进行过滤,得到合成例5的聚酯组合物。
[0134]
[比较合成例1]
[0135]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、160.35克的新戊二醇、555.00克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.21克的四异丙基钛酸酯作为触媒,在220℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在230℃下进行减压酯化反应,酯化反应共历时325分钟,且酯化终点的酸价为3.21mg koh/g。接着,将压力减至0托而在210℃下进行减压聚缩合反应,聚缩合反应共历时524分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土hyflo。接着,利用板框式过滤器进行过滤,得到比较合成例1的聚酯组合物。
[0136]
[比较合成例2]
[0137]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、561.23克的新戊二醇、208.13克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.22克的四异丙基钛酸酯作为触媒,在220℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在220℃下进行减压酯化反应,酯化反应共历时310分钟,且酯化终点的酸价为4.70mg koh/g。接着,将压力减至0托而在220℃下进行减压聚缩合反应,聚缩合反应共历时492分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土hyflo。接着,利用板框式过滤器进行过滤,得到比较合成例2的聚酯组合物。
[0138]
[比较合成例3]
[0139]
在装备有搅拌器、温度计、蒸馏管及冷凝计的3升圆底烧瓶中,添加1000克的己二酸、641.41克的新戊二醇、138.75克的2-甲基-1,3-丙二醇、155.95克的2-乙基己醇、与274.02克的月桂酸。在流通氮气并进行搅拌的条件下,在150℃下进行常压酯化反应且开始脱水。接着,加入0.22克的四异丙基钛酸酯作为触媒,在220℃及氮气气氛下进行常压酯化反应。之后,将压力减至460托至200托,在225℃下进行减压酯化反应,酯化反应共历时271分钟,且酯化终点的酸价为4.98mg koh/g。接着,将压力减至0托而在230℃下进行减压聚缩合反应,聚缩合反应共历时300分钟。之后,使产物降温至105℃,并添加0.05重量%(以产物的总重量计)的硅藻土hyflo。接着,利用板框式过滤器进行过滤,得到比较合成例3的聚酯组合物。
[0140]
3.2.2.聚酯组合物的性质分析
[0141]
依照前文所记载方法测定合成例1至5及比较合成例1至3的gc图谱、gc-ms图谱、酸价、羟基价、与黏度,并分别计算表示第一组分、第二组分与第三组分的层析峰的面积,将结果纪录于表1。
[0142]
表1:聚酯组合物的性质
[0143][0144]
3.3.热塑性高分子材料的制备与性质分析
[0145]
3.3.1.热塑性高分子材料的制备
[0146]
[实施例1]
[0147]
将100重量份的聚氯乙烯(型号:s-70,购自台塑)、50重量份的合成例1的聚酯组合物、2重量份的环氧化大豆油(型号:b-22,购自长春石油化学)、及2.5重量份的钙锌系安定剂(型号:mark37w,购自长江化学)置于试验用混合机(型号:mt22615,购自益宗精机)中,在172℃下混练7分钟,得到实施例1的热塑性高分子材料。
[0148]
[实施例2]
[0149]
将100重量份的聚氯乙烯s-70、50重量份的合成例2的聚酯组合物、2重量份的环氧化大豆油b-22、及2.5重量份的钙锌系安定剂mark 37w置于试验用混合机mt22615中,在172℃下混练7分钟,得到实施例2的热塑性高分子材料。
[0150]
[实施例3]
[0151]
将100重量份的聚氯乙烯s-70、50重量份的合成例3的聚酯组合物、2重量份的环氧化大豆油b-22、及2.5重量份的钙锌系安定剂mark 37w置于试验用混合机mt22615中,在172℃下混练7分钟,得到实施例3的热塑性高分子材料。
[0152]
[实施例4]
[0153]
将100重量份的聚氯乙烯s-70、50重量份的合成例4的聚酯组合物、2重量份的环氧化大豆油b-22、及2.5重量份的钙锌系安定剂mark 37w置于试验用混合机mt22615中,在172℃下混练7分钟,得到实施例4的热塑性高分子材料。
[0154]
[实施例5]
[0155]
将100重量份的聚氯乙烯s-70、50重量份的合成例5的聚酯组合物、2重量份的环氧化大豆油b-22、及2.5重量份的钙锌系安定剂mark 37w置于试验用混合机mt22615中,在172℃下混练7分钟,得到实施例5的热塑性高分子材料。
[0156]
[比较实施例1]
[0157]
将100重量份的聚氯乙烯s-70、50重量份的比较合成例1的聚酯组合物、2重量份的环氧化大豆油b-22、及2.5重量份的钙锌系安定剂mark 37w置于试验用混合机mt22615中,在172℃下混练7分钟,得到比较实施例1的热塑性高分子材料。
[0158]
[比较实施例2]
[0159]
将100重量份的聚氯乙烯s-70、50重量份的比较合成例2的聚酯组合物、2重量份的环氧化大豆油b-22、及2.5重量份的钙锌系安定剂mark 37w置于试验用混合机mt22615中,在172℃下混练7分钟,得到比较实施例2的热塑性高分子材料。
[0160]
[比较实施例3]
[0161]
将100重量份的聚氯乙烯s-70、50重量份的比较合成例3的聚酯组合物、2重量份的环氧化大豆油b-22、及2.5重量份的钙锌系安定剂mark 37w置于试验用混合机mt22615中,在172℃下混练7分钟,得到比较实施例3的热塑性高分子材料。
[0162]
3.3.2.热塑性高分子材料的性质分析(一)
[0163]
依照前文所记载方法测定实施例1至5及比较实施例1至3的热塑性高分子材料的初始拉伸强度、初始伸长率、初始伸长率100%模数、挥发率、拉伸残率、伸长率残率、及耐热老化后伸长率100%模数,将结果纪录于表2。
[0164]
表2:实施例及比较实施例的热塑性高分子材料的性质
[0165][0166]
如表2所示,使用本发明聚酯组合物作为可塑剂的热塑性高分子材料具有良好的初始拉伸强度、初始伸长率及100%模数等性质,且经耐热老化后仍具有良好的拉伸残率、伸长率残率及100%模数等性质。相较之下,如表2所示,使用非本发明聚酯组合物作为可塑剂的热塑性高分子材料无法同时具有良好的初始拉伸强度、初始伸长率及100%模数等性质,以及经耐热老化后的合宜的初始拉伸强度、初始伸长率及100%模数等性质。具体言之,如比较实施例1所示,若表示第一组分的层析峰的面积占比高于指定范围,则热塑性高分子材料在耐热老化后的伸长率残率不佳。如比较实施例2及比较实施例3所示,若表示第一组分的层析峰的面积占比低于指定范围,则热塑性高分子材料的初始伸长率100%模数不佳,会导致可塑化效率降低。
[0167]
3.3.3.热塑性高分子材料的性质分析(二)
[0168]
依照前文所记载方法选定实施例2至5与比较例1及3的热塑性高分子材料进行耐移行性测试,并将结果纪录于表3。
[0169]
表3:实施例2至5与比较例1及3的耐移行性测试结果
[0170][0171]
如表3所示,使用本发明聚酯组合物作为可塑剂的热塑性高分子材料的耐移行性佳,也即,聚酯组合物不易迁移或浸蚀到树脂板表面而造成损伤。相较之下,如表3所示,使
用非本发明聚酯组合物作为可塑剂的热塑性高分子材料的耐移行性不良,具体言之,比较例1及3显示,若作为可塑剂的聚酯组合物中表示第二组分的层析峰的面积占比落在指定范围外,则聚酯组合物的耐移行性不佳,在树脂板的表面可观察到明显的压痕。
[0172]
上述实施例仅为例示性说明本发明的原理及其功效,并阐述本发明的技术特征,而非用于限制本发明的保护范畴。任何本领域技术人员在不违背本发明的技术原理及精神下,可轻易完成的改变或安排,均属本发明所主张的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1