一种含氮化合物、其制备方法及应用与流程
1.本发明涉及一种含氮化合物、其制备方法及应用。
背景技术:
2.盐酸纳布啡,化学名(-)-17-(环丁烷甲基)-4,5α-环氧吗啡-3,6α,14-羟基盐酸盐,于1979年首次批准在美国上市,用于治疗中、重度疼痛及术后疼痛。2015年,宜昌人福药业盐酸纳布啡注射液批准上市,作为复合麻醉时诱导麻醉的辅助用药。其结构式如下所示:
[0003][0004]
文献报道(郑华章,李杰,张迎庆.盐酸纳布啡的合成.化学与生物工程,2007,24(9),19-21.)纳布啡的合成方法为:以蒂巴因为起始原料,经氧化得到14-羟基可待因酮(3);进行催化氢化制得中间体4;乙酸酐中乙酰化得到中间体5;与溴化氰反应得到6;用三溴化硼脱3位氧上的甲基得7;硫酸水解得8;与溴甲基环丁烷进行缩合得到中间体9;用硼氢化钠还原6位酮羰基制得纳布啡;最后加盐酸得到盐酸纳布啡。工艺路线如下所示:
[0005]
[0006]
文献对盐酸纳布啡有关物质的研究较少,而有关物质的研究关系着盐酸纳布啡原料药的稳定性以及用药安全性。因此,本领域亟需研究盐酸纳布啡的有关物质。
技术实现要素:
[0007]
本发明所要解决的技术问题是现有文献对盐酸纳布啡有关物质的研究较少,为此,本发明提供了一种含氮化合物、其制备方法及应用。该化合物有助于盐酸纳布啡的质量控制,提升用药安全性。同时,以蒂巴因为原料,经三步反应即可制得如式i所示的含氮化合物,该制备方法较为高效。
[0008]
本发明提供了一种如式i所示的含氮化合物或其盐;
[0009][0010]
在某一方案中,所述的如式i所示的含氮化合物或其盐可为用于物质y质量控制的如式i所示的含氮化合物或其盐;所述的物质y为纳布啡、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物。所述的纳布啡的药学上可接受的盐的溶剂合物可为纳布啡盐酸盐倍半水合物
[0011][0012]
在某一方案中,所述的如式i所示的含氮化合物或其盐可为用于物质y质量控制、作为对照品的如式i所示的含氮化合物或其盐;所述的物质y为纳布啡、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物。所述的纳布啡的药学上可接受的盐的溶剂合物可为纳布啡盐酸盐倍半水合物。
[0013]
在某一方案中,所述的如式i所示的含氮化合物或其盐可为用作物质y的有关物质的如式i所示的含氮化合物或其盐;所述的物质y为纳布啡、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物。所述的纳布啡的药学上可接受的盐的溶剂合物可为纳布啡盐酸盐倍半水合物。
[0014]
在某一方案中,所述的“如式i所示的含氮化合物的盐”中的“盐”可为药品质量控制领域常规使用的盐,例如与酸形成的盐。所述的酸可为本领域常规的酸,例如盐酸、氢溴酸、氢碘酸、硫酸、硝酸或磷酸。
[0015]
在某一方案中,所述的如式i所示的含氮化合物的盐可为
[0016]
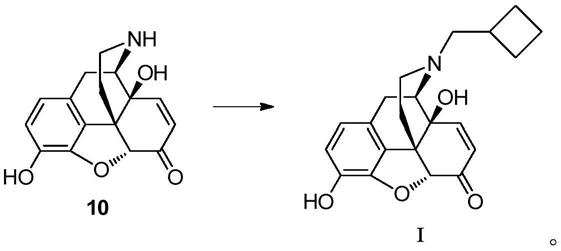
本发明提供了一种如式i所示的含氮化合物的制备方法,其包括下述步骤:在极性溶剂中,将化合物10与环丁基甲醛进行缩合反应,再经三乙酰氧基硼氢化钠还原,得到如式i所示的含氮化合物即可;
[0017][0018]
在所述的如式i所示的含氮化合物的制备方法中,所述的极性溶剂可为本领域该类反应常规的极性溶剂,例如二甲亚砜。
[0019]
在所述的如式i所示的含氮化合物的制备方法中,所述的极性溶剂的用量可为本领域该类反应常规的用量。例如,所述的化合物10与所述的极性溶剂的质量体积比为0.05g/ml~0.2g/ml;又例如,所述的化合物10与所述的极性溶剂的质量体积比为0.1g/ml。
[0020]
在所述的如式i所示的含氮化合物的制备方法中,所述的环丁基甲醛的用量可为本领域该类反应常规的用量。例如,所述的化合物10与所述的环丁基甲醛的摩尔比为1:1~1:2;又例如,所述的化合物10与所述的环丁基甲醛的摩尔比为1:1.2。
[0021]
在所述的如式i所示的含氮化合物的制备方法中,所述的化合物10与所述的三乙酰氧基硼氢化钠的摩尔比可为1:1~1:2,又可为1:1.3。
[0022]
在所述的如式i所示的含氮化合物的制备方法中,所述的缩合反应的温度可为本领域该类反应常规的温度,例如20℃~60℃,又例如20℃~35℃。
[0023]
在所述的如式i所示的含氮化合物的制备方法中,所述的缩合反应以化合物10的含量不再减少为止。其反应时间可为1h。
[0024]
在所述的如式i所示的含氮化合物的制备方法中,所述的还原反应的温度可为本领域该类反应常规的温度,例如20℃~60℃,又例如20℃~35℃。
[0025]
在所述的如式i所示的含氮化合物的制备方法中,所述的还原反应以如式i所示的含氮化合物不再增加为止。其反应时间可为1.5h。
[0026]
所述的如式i所示的含氮化合物的制备方法可进一步包括下述步骤:在卤代烃类溶剂中,在碱的存在下,将化合物10-a与氯甲酸乙酯进行取代反应,再在酸的存在下水解,得到所述的化合物10即可;
[0027][0028]
在所述的如式i所示的含氮化合物的制备方法中,所述的卤代烃类溶剂可为本领域该类反应常规的卤代烃类溶剂,例如氯仿。
[0029]
在所述的如式i所示的含氮化合物的制备方法中,所述的卤代烃类溶剂的用量可为本领域该类反应常规的用量。例如,所述的化合物10-a与所述的卤代烃类溶剂的摩尔体积比为1mol:(2~5)l;又例如,所述的化合物10-a与所述的卤代烃类溶剂的摩尔体积比为1mol:3l
[0030]
在所述的如式i所示的含氮化合物的制备方法中,所述的碱可为本领域该类反应常规的碱,例如无机碱,又例如碳酸钾。
[0031]
在所述的如式i所示的含氮化合物的制备方法中,所述的碱的用量可为本领域该类反应常规的用量。例如,所述的化合物10-a与所述的碱的摩尔比为1:(2~8);又例如,所述的化合物10-a与所述的碱的摩尔比为1:6
[0032]
在所述的如式i所示的含氮化合物的制备方法中,所述的氯甲酸乙酯的用量可为本领域该类反应常规的用量。例如,所述的化合物10-a与所述的氯甲酸乙酯的摩尔比为1:(2~8);又例如,所述的化合物10-a与所述的氯甲酸乙酯的摩尔比为1:6。
[0033]
在所述的如式i所示的含氮化合物的制备方法中,所述的酸可为本领域该类反应常规的酸,例如硫酸。
[0034]
在所述的如式i所示的含氮化合物的制备方法中,所述的酸的用量可为本领域该类反应常规的用量。例如,所述的化合物10-a与所述的酸的摩尔比为1:(5~15);又例如,所述的化合物10-a与所述的酸的摩尔比为1:10。
[0035]
在所述的如式i所示的含氮化合物的制备方法中,所述的取代反应的温度可为本领域该类反应常规的温度,例如50℃~60℃,又例如60℃。
[0036]
在所述的如式i所示的含氮化合物的制备方法中,所述的取代反应以化合物10-a的含量不再减少为止。其反应时间可为10h。
[0037]
在所述的如式i所示的含氮化合物的制备方法中,所述的水解反应的温度可为本领域该类反应常规的温度,例如90℃~100℃,又例如100℃。
[0038]
在所述的如式i所示的含氮化合物的制备方法中,所述的水解反应以化合物10的含量不再增加为止。其反应时间可为2-4h。
[0039]
所述的如式i所示的含氮化合物的制备方法可进一步包括下述步骤:在甲磺酸中,在蛋氨酸的存在下,将化合物3进行去甲基反应,得到所述的化合物10-a即可;
[0040][0041]
在所述的如式i所示的含氮化合物的制备方法中,所述的甲磺酸的用量可为本领域该类反应常规的用量。例如,所述的化合物3与所述的甲磺酸的质量体积比为1g:(5~15)ml;又例如,所述的化合物3与所述的甲磺酸的质量体积比为1g:10ml。
[0042]
在所述的如式i所示的含氮化合物的制备方法中,所述的蛋氨酸的用量可为本领域该类反应常规的用量。例如,所述的化合物3与所述的蛋氨酸的摩尔比为1:(1~3);又例如,所述的化合物3与所述的蛋氨酸的摩尔比为1:2。
[0043]
在所述的如式i所示的含氮化合物的制备方法中,所述的去甲基反应的温度可为本领域该类反应常规的温度,例如50℃~60℃,又例如55℃。
[0044]
在所述的如式i所示的含氮化合物的制备方法中,所述的去甲基反应以化合物3的含量不再减少为止。其反应时间可为7天。
[0045]
本发明还提供了一种上述的如式i所示的含氮化合物的盐的制备方法,其包括下述步骤:
[0046]
(1)按照上述的如式i所示的含氮化合物的制备方法,制得如式i所示的含氮化合物;
[0047]
(2)在溶剂中,将步骤(1)制得的如式i所示的含氮化合物与酸进行成盐反应,得到如式i所示的含氮化合物的盐即可。
[0048]
在所述的如式i所示的含氮化合物的盐的制备方法中,步骤(2)里,所述的溶剂可为成盐反应常规的溶剂,例如醇类溶剂,又例如乙醇。
[0049]
在所述的如式i所示的含氮化合物的盐的制备方法中,步骤(2)里,所述的溶剂的用量可为本领域该类成盐反应常规的用量。例如,所述的溶剂与所述的如式i所示的含氮化合物的体积摩尔比可为(5~10)ml:1mol;又例如,所述的溶剂与所述的如式i所示的含氮化合物的体积摩尔比可为6ml:1mol。
[0050]
在所述的如式i所示的含氮化合物的盐的制备方法中,步骤(2)里,所述的酸可为成盐反应常规的酸,例如hcl。所述的hcl可以以盐酸水溶液的形式使用。所述的盐酸水溶液可为12mol/l盐酸水溶液。
[0051]
在所述的如式i所示的含氮化合物的盐的制备方法中,步骤(2)里,所述的酸的用量可为本领域该类成盐反应常规的用量。例如,所述的酸与所述的如式i所示的含氮化合物的摩尔比可为(1.0~1.2):1;又例如,所述的酸与所述的如式i所示的含氮化合物的摩尔比可为1.1:1。
[0052]
本发明还提供了一种化合物10的制备方法,其包括下述步骤:在卤代烃类溶剂中,在碱的存在下,将化合物10-a与氯甲酸乙酯进行取代反应,再在酸的存在下水解,得到化合物10即可;
[0053][0054]
所述的取代反应、水解反应的反应条件如上所述。
[0055]
所述的化合物10的制备方法可进一步包括下述步骤:在甲磺酸中,在蛋氨酸的存在下,将化合物3进行去甲基反应,得到所述的化合物10-a即可;
[0056][0057]
所述的去甲基反应的反应条件如上所述。
[0058]
本发明还提供了一种化合物10-a的制备方法,其包括下述步骤:在甲磺酸中,在蛋氨酸的存在下,将化合物3进行去甲基反应,得到化合物10-a即可;
[0059][0060]
所述的去甲基反应的反应条件如上所述。
[0061]
本发明还提供了一种物质x作为物质y的有关物质的应用;
[0062]
所述的物质x为上述的如式i所示的含氮化合物或其盐;所述的物质y为纳布啡、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物。
[0063]
在某一方案中,所述的纳布啡的药学上可接受的盐的溶剂合物可为纳布啡盐酸盐倍半水合物。
[0064]
本发明还提供了一种物质x在物质y的质量控制中作为对照品的应用;
[0065]
所述的物质x为上述的如式i所示的含氮化合物或其盐;所述的物质y为纳布啡、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物。
[0066]
在某一方案中,所述的纳布啡的药学上可接受的盐的溶剂合物可为纳布啡盐酸盐倍半水合物。
[0067]
本发明还提供了一种药物组合,其包括物质x和物质y;
[0068]
所述的物质x为上述的如式i所示的含氮化合物或其盐;所述的物质y为纳布啡、其药学上可接受的盐、其溶剂合物或其药学上可接受的盐的溶剂合物。
[0069]
在某一方案中,所述的药物组合由所述的物质x和所述的物质y组成。
[0070]
在某一方案中,所述的物质x在所述的药物组合中的质量百分比可为1%以下,又
可为0.5%以下,还可为0.1%以下,更可为0.01%以下。
[0071]
在某一方案中,所述的物质x为在某一方案中,所述的物质x为
[0072]
在某一方案中,所述的物质y里,所述的纳布啡的药学上可接受的盐的溶剂合物可为纳布啡盐酸盐倍半水合物。
[0073]
术语“药学上可接受的盐”是指纳布啡与相对无毒的、药学上可接受的酸或碱制备得到的盐。纳布啡中的相对酸性的功能团可以通过在纯的溶液或合适的惰性溶剂中用足够量的药学上可接受的碱与纳布啡的中性形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括但不限于:锂盐、钠盐、钾盐、钙盐、铝盐、镁盐、锌盐、铋盐、铵盐、二乙醇胺盐。纳布啡中的相对碱性的官能团可以通过在纯的溶液或合适的惰性溶剂中用足够量的药学上可接受的酸与纳布啡的中性形式接触的方式获得酸加成盐。所述的药学上可接受的酸包括无机酸,所述无机酸包括但不限于:盐酸、氢溴酸、氢碘酸、硝酸、碳酸、磷酸、亚磷酸、硫酸等。所述的药学上可接受的酸包括有机酸,所述有机酸包括但不限于:乙酸、丙酸、草酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、反丁烯二酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、水杨酸、酒石酸、甲磺酸、异烟酸、酸式柠檬酸、油酸、单宁酸、泛酸、酒石酸氢、抗坏血酸、龙胆酸、富马酸、葡糖酸、糖酸、甲酸、乙磺酸、双羟萘酸(即4,4
’‑
亚甲基-双(3-羟基-2-萘甲酸))、氨基酸(例如谷氨酸、精氨酸)等。具体可参见berge et al.,"pharmaceutical salts",journal of pharmaceutical science 66:1-19(1977)、或、handbook of pharmaceutical salts:properties,selection,and use(p.heinrich stahl and camille g.wermuth,ed.,wiley-vch,2002)。
[0074]
术语“溶剂合物”是指纳布啡与化学计量或非化学计量的溶剂结合形成的物质。所述的溶剂包括但不限于:水、甲醇、乙醇等。
[0075]
术语“药学上可接受的盐的溶剂合物”中的“药学上可接受的盐”和“溶剂合物”如上所述,是指纳布啡与1、与相对无毒的、药学上可接受的酸或碱制备得到的2、与化学计量或非化学计量的溶剂结合形成的物质。所述的“药学上可接受的盐的溶剂合物”包括但不限于纳布啡盐酸盐倍半水合物。
[0076]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0077]
本发明所用试剂和原料均市售可得。
[0078]
本发明的积极进步效果在于:如式i所示的含氮化合物有助于盐酸纳布啡的质量控制,提升用药安全性。同时,以蒂巴因为原料,经三步反应即可制得如式i所示的含氮化合物,该制备方法较为高效。
具体实施方式
[0079]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0080]
实施例1
[0081]
按照wo 2015066443a1实施例1记载的方法,以去甲羟吗啡酮为原料制得纳布啡。在表1所示的hplc条件下检测制得的纳布啡,于保留时间为20.1min处发现一色谱峰。但其含量在0.1%以下,且与(保留时间为20.8min)、纳布啡(保留时间为19.2min)接近,分离难度很大。
[0082]
表1
[0083][0084]
为分离上述保留时间为20.1min的物质,对原料去甲羟吗啡酮进行下述处理:取去甲羟吗啡酮,经过成盐纯化并在thf中热打浆,过滤,取滤液,旋蒸,重复上述操作3次,获得新的去甲羟吗啡酮。随后,新的去甲羟吗啡酮按照wo 2015066443a1实施例1记载的方法,制
得纳布啡。取纳布啡20g,经过硅胶柱层析(dcm:meoh=20:1),获得40mg产品。采用表1的所示的hplc条件检测,保留时间为20.1min;该产品的鉴定数据如下:
[0085]
ms(esi
+
):m/z,(354.16[m+h]
+
);
[0086]1h nmr(400mhz,cd3od)δ6.94(d,j=10.2hz,1h),6.69(s,2h),6.16(d,j=10.2hz,1h),4.79(s,1h),3.81(d,j=6.1hz,1h),3.54-3.39(m,2h),3.24-3.13(m,2h),3.03(dd,j=19.8,6.3hz,1h),2.89(td,j=12.7,3.4hz,1h),2.83-2.70(m,2h),2.35-2.13(m,2h),2.11-1.90(m,4h),1.89-1.81(m,1h).
[0087]
13
c nmr(101mhz,cd3od)δ195.29,147.63,144.53,141.66,134.59,130.17,122.01,121.69,119.98,87.23,68.45,62.90,59.07,48.60,47.59,32.03,28.14,28.07,26.58,24.22,19.41.
[0088]
结合纳布啡的制备方法,确证其结构为
[0089]
实施例2
[0090][0091]
步骤(1):14-羟基可待因酮(化合物3)的合成
[0092]
蒂巴因(5.0g,16.1mmol),85%甲酸2.5ml,2%稀硫酸7.5ml,加入到反应瓶中,搅拌溶解。冷却到5℃,滴入30%过氧化氢(3ml,29.3mmol),在40℃搅拌反应6.5h,然后加入活性炭4g、丙酮-水(1:1)50ml,继续搅拌脱色1小时。趁热过滤,滤液在冰水浴冷却下用氨水调ph=6,加入少量亚硫酸氢钠,继续用氨水调ph=8,析出白色沉淀,冷藏,过滤,分别用少量蒸馏水和乙醇洗涤,干燥,得白色粉末状固体4.0g,收率79.5%,纯度98.8%。1h nmr(400mhz,cdcl3)δ6.69(d,j=8.2hz,1h),6.62(d,j=9.6hz,2h),6.17(d,j=10.1hz,1h),4.70(s,1h),3.84(s,3h),3.24(d,j=18.7hz,1h),3.04(d,j=5.9hz,1h),2.58
–
2.49(m,2h),2.45(s,3h),2.41(dd,j=12.5,5.0hz,1h),2.28(td,j=11.8,3.6hz,1h),1.68(dd,j=12.6,2.9hz,1h).
13
c nmr(101mhz,cdcl3)δ194.28,147.50,144.38,142.69,134.63,130.54,125.07,119.61,115.16,87.11,67.81,64.18,56.87,46.65,45.17,42.59,29.49,22.41.hrms(esi)m/z 314.1394(计算值c
18h20
no4,314.1392[m+h]
+
).
[0093]
步骤(2):7,8-二氢-4,5α-环氧吗啡喃-6-酮-3,14-二醇(化合物10)的合成
[0094]
将蛋氨酸(3.8g,25.5mmol)的甲磺酸(40ml)溶液加入反应釜中,加入化合物3(4.0g,12.7mmol),加热至55℃,反应七天。用甲醇(60ml)和水(120ml)淬灭,控制温度在20℃以下,用20%naoh调ph至9,用二氯甲烷和乙酸乙酯萃取,旋蒸得到黄色固体10-a。将所得黄色固体溶解在氯仿中(30ml),加入碳酸钾(10.5g,76.6mmol),然后加入氯甲酸乙酯(8.3g,76.6mmol)的氯仿(10ml)溶液。加热至回流,搅拌10小时,冷却至室温,过滤,滤液浓缩得油状物,加入稀硫酸{浓硫酸(12.5g,125mmol),加水38ml},氮气保护,加热水解加热至回流反应2-4h,反应毕,降温至30℃左右,搅拌滴加氨水,调ph=8-9,冰水浴下搅拌2h,抽滤,水洗,所得固体经柱层析纯化得到化合物(10)1.82g类白色固体,收率50.3%,纯度99.1%。1h nmr(400mhz,cd3od)δ6.90(d,j=10.2hz,1h),6.68(q,j=8.2hz,2h),6.15(d,j=10.2hz,1h),4.77(s,1h),3.95(dd,j=4.9,1.8hz,1h),3.27-3.14(m,3h),2.93(td,j=13.3,4.3hz,1h),2.73(td,j=13.3,5.0hz,1h),1.84(dd,j=13.4,3.6hz,1h).
13
c nmr(101mhz,cd3od)δ195.48,147.87,144.65,141.66,134.46,130.31,122.42,121.55,119.97,87.47,67.45,58.25,47.81,38.49,28.86,26.96.ms(esi+):m/z,(286.08[m+h]
+
).
[0095]
步骤(3):17-(环丁烷甲基)-7,8-二氢-4,5α-环氧吗啡喃-6-酮-3,14-二醇(式i化合物)的合成
[0096]
化合物(10)(1.0g,3.5mmol)溶于dmso(10ml)中,加入环丁基甲醛(0.35g,4.2mmol),室温(20-35℃)搅拌1h后,加入三乙酰氧基硼氢化钠(nabh(oac)3)(0.96g,4.6mmol),反应1.5h后,加入水(10ml),浓氨水调ph至9,室温下搅拌2h,过滤,水洗,得类白色固体0.9g,收率72.3%,纯度97.3%。1h nmr(400mhz,cd3od)δ6.94(d,j=10.2hz,1h),6.69(s,2h),6.16(d,j=10.2hz,1h),4.79(s,1h),3.81(d,j=6.1hz,1h),3.54-3.39(m,2h),3.24-3.13(m,2h),3.03(dd,j=19.8,6.3hz,1h),2.89(td,j=12.7,3.4hz,1h),2.83-2.70(m,2h),2.35-2.13(m,2h),2.11-1.90(m,4h),1.89-1.81(m,1h).
13
c nmr(101mhz,cd3od)δ195.29,147.63,144.53,141.66,134.59,130.17,122.01,121.69,119.98,87.23,68.45,62.90,59.07,48.60,47.59,32.03,28.14,28.07,26.58,24.22,19.41.ms(esi+):m/z,(354.16[m+h]
+
).
[0097]
上述的鉴定数据与实施例1的鉴定数据相同,进一步确证实施例1分离得到的产物为
[0098]
步骤(4):17-(环丁烷甲基)-7,8-二氢-4,5α-环氧吗啡喃-6-酮-3,14-二醇一盐酸盐(式i化合物的一盐酸盐)的合成
[0099]
将0.5g式i化合物(1.4mmol)加入到乙醇中(8ml),滴加12mol/l盐酸0.13ml(1.6mmol),搅拌1h,旋干溶剂,得式i化合物的一盐酸盐0.54g(收率98.0%),白色固体。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1