一种制备喜树碱类和伊文思蓝偶联的两亲性化合物的方法与流程
1.本发明属于化合物合成技术领域,具体涉及一种喜树碱类和伊文思蓝形成的小分子两亲性药物(简称eb
‑
ss
‑
cpt)的制备方法。
背景技术:
2.物质的自组装是推动自然界中生物结构和功能进化的最有趣的过程。在过去的几十年里,自组装在为生物医学应用量身定制的多种纳米材料的研发中发挥了重要作用。由于生物屏障的存在,小分子治疗药物在体内溶解性差,药效学和药代动力学不够理想,导致药物传递和利用效率低下。纳米医学的出现使治疗药物可以以纳米材料作为载体,并可以根据不同的参数进行优化,以改善治疗效果。
3.现有技术中,喜树碱类和伊文思蓝通过连接结构偶联形成的小分子化合物具有两亲性,记作“eb
‑
cpt”。研究发现,该化合物可以在溶剂中自发地自组装形成胶束样颗粒,并具有一定的抗癌活性,可以作为理想的药物递送载体。但是现有的eb
‑
cpt化合物制备方法副反应较多,反应效率和反应收率都不够理想,纯化难度较大,不利于规模化工业生产。
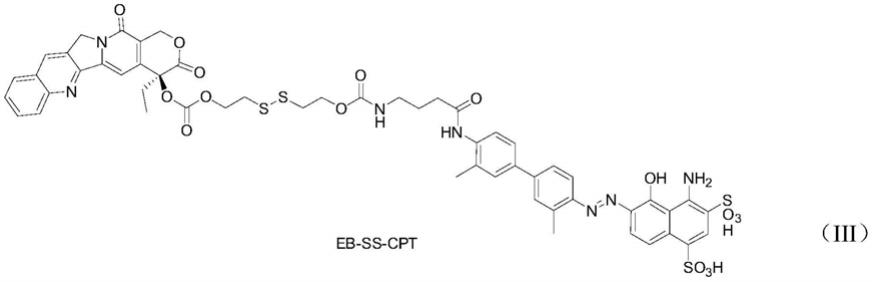
4.因此有必要对eb
‑
cpt的合成方法进行优化,以促进eb
‑
cpt的规模化生产,进一步满足纳米抗癌药物临床应用的需要。
技术实现要素:
5.鉴于上述背景,本发明目的在于:提供一种喜树碱类和伊文思蓝偶联的两亲性化合物(eb
‑
cpt)的新的制备方法,使其具有更高的反应效率、更少的副反应和更高的收率,总体更适合工业化生产,且制得的eb
‑
cpt化合物具有更理想的自组装性能。
6.本发明的上述目的通过以下技术方案实现:
7.提供一种制备喜树碱类和伊文思蓝偶联的两亲性化合物的方法,包括以下步骤:
8.步骤i:喜树碱或其衍生物与羰基化试剂在有机溶剂中反应,后加入生成中间体f;其中n为0
‑
10的整数,优选为0
‑
5的整数,更优选为0
‑
3的整数;
9.步骤ii:步骤i所得中间体f的羟基与氯甲酸酯类化合物发生酰化反应生成中间体g;
10.步骤iii:邻连甲苯胺与在偶联试剂存在下发生酰胺键偶联缩合生产中间体d;其中m为0
‑
10的整数,优选为0
‑
5的整数,更优选为0
‑
3的整数;
11.步骤iv:步骤iii所得的中间体d与1
‑
氨基
‑8‑
萘酚
‑
2,4
‑
二磺酸单钠盐发生重氮化偶联反应生成中间体e;
12.步骤v:步骤iv所得的中间体e脱除保护基得到中间体c;
13.步骤vi:步骤v所得的中间体c与步骤ii得到的中间体g在有机溶剂中发生亲核取代反应生成eb
‑
ss
‑
cpt,即喜树碱类和伊文思蓝通过二硫键及氨基甲酸酯键偶联的两亲性
化合物。
14.本发明所述的制备方法中,步骤i所述的喜树碱或其衍生物选自以下式(i)或式(ii)所示的任意一种:
[0015][0016]
本发明所述的制备方法中,步骤i所述的羰基化试剂可以是光气、二光气、三光气或羰基二咪唑等羰基化试剂中的任意一种。
[0017]
本发明所述的制备方法中,步骤i所述的有机溶剂可以是二氯甲烷、三氯甲烷、四氢呋喃、乙酸乙酯、n,n'
‑
二甲基乙酰胺或1,2
‑
二氯乙烷中的任意一种。
[0018]
本发明所述的制备方法中,步骤ii所述的氯甲酸酯类化合物可以是氯甲酸
‑4‑
硝基苯酯或2,2,2
‑
三氯氯甲酸乙酯。
[0019]
本发明所述的制备方法中,步骤iii所述的偶联试剂可以是2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸盐(hatu)、o
‑
苯并三氮唑
‑
四甲基脲六氟磷酸盐(hbtu)、6
‑
氯苯并三氮唑
‑
1,1,3,3
‑
四甲基脲六氟磷酸酯(hctu)、1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edci)、六氟磷酸苯并三唑
‑1‑
氧基三(二甲氨基)磷(bop)或六氟磷酸苯并三唑
‑1‑
基
‑
氧基三吡咯烷基磷(pybop)中的任意一种。
[0020]
本发明所述的制备方法中,步骤v所述的脱除保护基可以使用以下溶剂处理中间体e:三氟乙酸、乙二硫醇和苯甲硫醚以(10
‑
100):(0
‑
90):(0
‑
90)的体积比混合得到的混合溶剂。
[0021]
本发明所述的制备方法中,步骤vi所述的有机溶剂可以是n,n'
‑
二甲基甲酰胺、n,n'
‑
二甲基乙酰胺或n
‑
甲基吡咯烷酮中的任意一种。
[0022]
本发明优选的一种实施方式中,所述制备方法包括以下步骤:
[0023]
步骤i:喜树碱与羰基化试剂在有机溶剂中反应,后加入双(2
‑
羟乙基)硫化物生成中间体f;
[0024]
步骤ii:步骤i所得的中间体f的羟基与氯甲酸
‑4‑
硝基苯酯发生酰化反应生成中间体g;
[0025]
步骤iii:邻连甲苯胺与n
‑
(叔丁氧羰基)
‑4‑
氨基丁酸在偶联试剂存在下发生酰胺键偶联缩合生产中间体d;
[0026]
步骤iv:步骤iii所得的中间体d与1
‑
氨基
‑8‑
萘酚
‑
2,4
‑
二磺酸单钠盐发生重氮化偶联反应生成中间体e;
[0027]
步骤v:步骤iv所得的中间体e脱除保护基得到中间体c;
[0028]
步骤vi:步骤v所得的中间体c与步骤ii得到的中间体g在有机溶剂中发生亲核取代反应生成eb
‑
ss
‑
cpt
‑
iii,即结构如下式(iii)所示的喜树碱类和伊文思蓝通过二硫键及氨基甲酸酯键偶联的两亲性化合物。
[0029][0030]
所述eb
‑
ss
‑
cpt
‑
iii的合成路线如下:
[0031]
[0032][0033]
与现有技术相比,本发明通过新的合成路线合成了由二硫键及氨基甲酸酯键将喜树碱类和伊文思蓝偶联在一起的eb
‑
cpt两亲性化合物。该方法合成步骤简单、反应效率高、副反应少、收率高,得到的产物极易纯化,总体上比现有的eb
‑
cpt合成方法更适合工业化生产,且制得的eb
‑
cpt化合物具有更理想的自组装性能。
附图说明
[0034]
图1是实施例2所述中间体e的质谱图。
[0035]
图2是实施例3所述中间体c的质谱图。
[0036]
图3是实施例5所述中间体g的质谱图。
[0037]
图4是实施例6所述产品eb
‑
ss
‑
cpt
‑
iii的质谱图。
[0038]
图5是实施例1所述中间体d的核磁氢谱图。
[0039]
图6是实施例2所述中间体e的核磁氢谱图。
[0040]
图7是实施例3所述中间体c的核磁氢谱图。
[0041]
图8是实施例5所述中间体g的核磁氢谱图。
[0042]
图9是实施例6所述产品eb
‑
ss
‑
cpt
‑
iii的核磁氢谱图。
具体实施方式
[0043]
下面结合实施例对本发明做进一步说明,但不因此而限定本发明的内容。
[0044]
实施例1:中间体d的制备
[0045]
取10.03g的n
‑
(叔丁氧羰基)
‑4‑
氨基丁酸、19.65g的hatu、15.67g的邻连甲苯胺、250ml的乙腈及41ml的dipea搅拌反应过夜,减压蒸馏除去乙腈,加入150ml的乙酸乙酯、150ml的纯化水,盐酸调节ph=5
‑
6,分液,有机相加入150ml的纯化水,盐酸调节ph=4
‑
5,分液,有机相无水硫酸钠干燥,蒸馏除去乙酸乙酯,得产品14.56g的中间体d,收率72%。其结构表征谱图见图5。
[0046]
实施例2:中间体e的制备
[0047]
取2.51g实施例1制备的中间体d、25ml的乙腈和12.5ml的2m的盐酸,降温至
‑5‑
5℃,滴加亚硝酸钠溶液(1.30g溶于13ml水中),另取一烧瓶加入2.00g的1
‑
氨基
‑8‑
萘酚
‑
2,4
‑
二磺酸单钠、2.11g碳酸氢钠和15ml水,降温至
‑5‑
5℃,将以上重氮盐滴加至上述溶液中,反应完毕后,加入乙酸乙酯,分液,水相减压蒸馏得中间体e(6.44g)。收率:100%(含有无机盐),理论[m+h]
+
=726.19;实测[m+h]
+
=726.19454。其结构表征谱图见图1和图6。
[0048]
实施例3:中间体c的制备
[0049]
取2.01g实施例2制备的中间体e,加入16ml三氟乙酸、2ml乙二硫醇和2ml苯甲硫醚的混合溶剂,反应2h,反应完毕,加入60ml乙酸乙酯,过滤,乙酸乙酯淋洗,烘干得1.53g中间体c。收率:100%(含有无机盐)理论[m
‑
h]
‑
=626.14;实测[m
‑
h]
‑
=626.13592。其结构表征谱图见图2和图7。
[0050]
实施例4:中间体f的制备
[0051]
称取40.14g双(2
‑
羟乙基)硫化物,加入thf溶解后称重得230.46g,加入适量活化好的分子筛密闭干燥。取4.00g喜树碱、6.30g的dmap加入1000ml的dcm,搅拌下滴加三光气的二氯甲烷溶液(三光气1.91g+200mldcm)。滴加完毕,称取197.69g上述干燥的双(2
‑
羟乙基)硫化物得四氢呋喃溶液,加至反应液中,室温搅拌过夜。反应液用600ml水洗、600ml的0.05m的盐酸洗、600ml水洗、600ml饱和食盐水洗,分液,有机相用无水硫酸钠干燥。过滤,旋干。
[0052]
固体加入100ml乙酸乙酯打浆,过滤,滤饼用20ml的ea淋洗。干燥,得5.43g固体,即中间体f。
[0053]
实施例5:中间体g的制备
[0054]
称取2.00g实施例4制备的中间体f和1.07g的dipea加入300ml的dcm搅拌溶解。称取0.76g的氯甲酸
‑4‑
硝基苯酯,加入100ml的dcm,溶解后滴加至上述含中间体f的反应液中反应过夜,补加1.08g的dipea和0.10g的dmap至反应液中。称取0.77g氯甲酸
‑
4硝基苯酯加入50ml的dcm,溶解后滴加至反应液中。反应40℃搅拌反应,无原料剩余后,反应液加入300ml水、300ml1m盐酸、300ml水和300ml饱和食盐水搅洗分液。有机相用无水硫酸钠干燥、过滤、旋干。柱层析分离产品得2.45g黄色固体,即化合物g。两步收率84%。理论[m+h]
+
=694.12;实测[m+h]
+
=694.11608。结构表征见图3和图8。
[0055]
实施例6:eb
‑
ss
‑
cpt
‑
iii的制备
[0056]
称取0.50g实施例3制备的化合物c、0.67g实施例5制备的化合物g、1.04g的dipea加入50ml的dmf,升温至30℃下搅拌反应过夜。量取400ml的dcm将反应液倒入dcm中,降温至0℃下搅拌。过滤,滤液40℃下真空干燥。得0.61g固体,即为本发明所述的喜树碱和伊文思蓝通过二硫键及氨基甲酸酯键偶联的两亲性化合物eb
‑
ss
‑
cpt
‑
iii。收率:54%。理论[m
‑
h]
‑
=1180.23;实测[m
‑
h]
‑
=1180.22139。结构表征谱图见图4和图9。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1