一种MPPR/POSS/GF复合协效无卤阻燃聚丙烯复合材料及其制备方法
一种mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料及其制备方法
技术领域
1.本发明属于单分子膨胀型阻燃剂技术领域,涉及一种mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料及其制备方法。
背景技术:
2.聚丙烯综合性能优良,应用广泛,但聚丙烯的全碳氢链结构使得pp极易燃烧,且燃烧时会产生大量有焰熔滴,释放大量的热量,极易传播火焰而引发火灾,危害人们的生命和财产安全,严重限制其在某些需要高阻燃性领域的应用。因此,为了拓宽聚丙烯的应用范围,研制新型阻燃pp复合材料十分迫切。根据目前聚丙烯用阻燃剂的发展和环保要求,卤系阻燃剂因燃烧时会释放出大量有毒烟雾和气体,严重危害人体健康和环保安全,其应用逐步受到限制,因此开发绿色、环保的无卤阻燃剂是聚丙烯阻燃的研究热点和发展方向。
3.膨胀型阻燃剂(ifr)由于具有低毒、低烟、阻燃效率较高等优点被认为是最有发展前途的无卤阻燃剂之一。传统研究较多的ifr一般以混合膨胀型阻燃剂为主,如由聚磷酸铵(app)、季戊四醇(per)和三聚氰胺(me)按一定比例复配而成的最简单混合ifr体系。但上述这种传统ifr体系热稳定性差,app和per容易吸湿析出,此外per也容易在加工和使用过程中迁移至基体表面,严重劣化pp的阻燃性能、力学性能和服役性能。因此,有必要研究和构建新的适于pp阻燃的膨胀阻燃体系。
4.植酸(pa),又名环己六醇六磷酸,分子式c6h
18
o
24
p6,主要存在于植物种子、根干和茎中,环境友好,磷含量高(高达28wt%)。目前,已有将植酸用于聚合物阻燃的文献报道,但将其应用于合成适于聚丙烯(pp)的无卤阻燃剂尤其是膨胀型阻燃剂的研究仍较少,且通常阻燃效率不佳,例如将植酸与哌嗪合成植酸盐阻燃pp时其氧指数仅能提高至25.5%。
5.而现有技术中,为了解决上述混合型膨胀阻燃剂的缺点,研究者合成了一种具酸源、炭源和气源三位一体的单分子膨胀型阻燃剂,并将其用于聚合物的阻燃。
6.中国授权发明“一种含磷
‑
氮
‑
碳聚合型膨胀型阻燃剂的制备方法及其用途”(授权公开号cn106519239b)公开了一种具纳米片状结构的含磷
‑
氮
‑
碳聚合型膨胀型阻燃剂的制备方法,其特征是:取植酸、三聚氰胺和溶剂,植酸与三聚氰胺的物质的量之比为1:3~1:15,溶剂的用量为每0.01mol三聚氰胺添加溶剂20~50ml;向装有温度计和搅拌器的反应容器中加入三聚氰胺,搅拌下加入溶剂,在温度20~40℃下加入植酸,搅拌反应20~50分钟,得反应液;将反应液倒入反应釜中,在温度70~120℃下反应24~56h,冷却、抽滤,固体物用乙醇洗涤后、再用水洗涤至ph为7,干燥,即制得。
7.该授权专利中是以植酸作为酸源和炭源,以三聚氰胺作为气源制备得到了简称为pama的膨胀型阻燃剂,并通过其说明书中应用例4公开了当pama为25wt%添加时阻燃聚丙烯(pp)达到了29.6%的氧指数,垂直燃烧性能ul
‑
94达到了v
‑
0等级。
8.但是,该专利是以植酸同时作为酸源和炭源,且通过较大的三聚氰胺添加比例(1:3~1:15)构成阻燃共聚单体。其中,植酸的具体化学结构式如下:
[0009][0010]
可明显看出植酸相对来说碳含量较低(10.9wt%,常用炭源季戊四醇的碳含量为44.1wt%),其作为炭源时不利于成炭反应的发生,并且膨胀型阻燃剂中三聚氰胺也因其碳含量较低通常无法作为合适的炭源。
[0011]
需要重点说明的是,膨胀型阻燃剂中炭源是形成泡沫炭质层的基础。聚合物燃烧时,酸源部分的主要作用是分解产生无机酸(通常为磷酸或偏磷酸),催化炭源(成炭剂)发生脱水反应形成炭层,同时偏磷酸还能促使聚合物脱水炭化,增强凝聚相阻燃效果,因此,一方面酸源中的磷含量高低对膨胀型阻燃剂的阻燃效果有很大影响,另一方面炭源也需具有较高的碳含量在燃烧时形成脱水炭层覆盖在阻燃材料的表面,有效隔绝基体与外界热量和氧气的传递,从而降低材料的可燃性。
[0012]
因此,虽然该授权专利技术方案在实验室测试条件下仍具有较佳的阻燃性能,但基于其阻燃剂组分构成的理论分析,pama总体碳含量低下,在基体材料点燃时,其炭化速度较慢,由气源分解产生的不燃气体可能将炭层顶起或吹破,不能形成有效的膨胀炭层结构隔绝基体与外界热氧的接触,在发生大面积着火时难以自熄,不能取得理想的阻燃效果。
[0013]
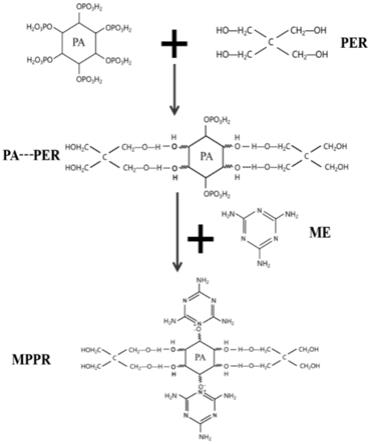
但是,若在该专利技术内容的基础中进一步添加使用充当炭源的组分(如季戊四醇)复配制备,如该授权专利背景技术中记载:
“……
在实际应用中往往需要和一些充当炭源的物质(如季戊四醇)复配使用才能具有较好的效果,同时需要较高的添加量。”又根据现有技术可知传统单分子膨胀型阻燃剂中聚磷酸铵、三聚氰胺、季戊四醇物质的量比例通常为2:1:1左右,因此在该专利技术内容上基础上的确需要添加较高添加量的季戊四醇,但此时植酸的添加量过低导致磷含量降低将极大的影响到阻燃性能。
[0014]
同时,又由于植酸、三聚氰胺、季戊四醇的酸源、气源、炭源体系为三组分体系,且三聚氰胺与季戊四醇都为多官能团化合物,都存在有多个基团能够与植酸中六个磷酸基团形成化学键,无法通过彼此间的化学结构确定最适宜的反应合成比例。因此为了提高使用植酸作为酸源的膨胀型阻燃剂的综合阻燃性能,势必需要针对酸源、气源、炭源三组分体系进行更深一步的探索与研究。
技术实现要素:
[0015]
本发明的目的在于克服上述的现有技术问题,提供一种mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料及其制备方法。制备所得阻燃聚丙烯复合材料阻燃效率高,力学强度高,可满足阻燃pp在高强度高模量应用场合的要求。
[0016]
为实现上述目的,本发明是采用由以下技术措施构成的技术方案来实现的。
[0017]
一种mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料,按重量份数计,主要包括
以下组分:
[0018][0019]
且总重量份数为100份;
[0020]
所述单分子膨胀型阻燃剂mppr,是由组分包括植酸、多元醇、三聚氰胺制备得到,且植酸、多元醇、三聚氰胺摩尔比为1:(2.4~2.6):(1.9~2.1);
[0021]
其中所述多元醇为季戊四醇、双季戊四醇和三季戊四醇中的至少一种。
[0022]
上述单分子膨胀型阻燃剂mppr的制备方法,包括以下步骤:
[0023]
(1)将植酸配置为植酸溶液;
[0024]
(2)将多元醇加入步骤(1)所得的植酸溶液,于95~100℃条件下搅拌反应至少1h;再加入三聚氰胺,继续于95~100℃条件下搅拌反应至少0.5h,即得混合溶液;
[0025]
(3)将步骤(2)所得混合溶液冷却后进行干燥至恒重,即得单分子膨胀型阻燃剂mppr。
[0026]
其中,上述制备方法中所用植酸、多元醇、三聚氰胺摩尔比为1:(2.4~2.6):(1.9~2.1),其中所述多元醇为季戊四醇、双季戊四醇和三季戊四醇中的至少一种。
[0027]
通常而言,植酸为易溶于水的液体,因此步骤(1)中将植酸配置为植酸溶液,为根据植酸的添加量选择适宜体积的去离子水进行溶解配置为植酸溶液,从而方便后续与多元醇、三聚氰胺在水溶液环境中的反应完全。为了更好地说明本发明,并提供一种可选择的技术方案,步骤(1)中将植酸配置为植酸溶液,是将植酸以0.125~0.25mol/l的摩尔浓度比例配置为植酸溶液。
[0028]
其中,步骤(2)中所述将多元醇加入步骤(1)所得的植酸溶液,于95~100℃条件下搅拌反应至少1h,通常可选择封闭条件或冷凝回流条件下搅拌反应,防止该温度条件下因蒸发导致的反应体系溶液浓度提高,从而导致的反应不完全。
[0029]
其中,步骤(2)中所述将多元醇加入步骤(1)所得的植酸溶液,于95~100℃条件下的封闭体系中搅拌反应至少1h,所述搅拌反应时间为实验室条件下的经验总结。通常而言,实验室条件下,植酸的添加量在不高于1mol的情况时,加入多元醇,于95~100℃条件下的封闭体系中搅拌反应1~2h即可。在实际工业化生产中,本领域技术人员可根据实际情况及工业放大效应,选择适宜的搅拌反应时间,以达到植酸与多元醇的充分反应。
[0030]
其中,步骤(2)中所述加入三聚氰胺,继续于95~100℃条件下的开放体系中搅拌反应至少0.5h,所述搅拌反应时间为实验室条件下的经验总结。通常而言,实验室条件下,植酸的添加量在不高于1mol的情况时,加入多元醇,于95~100℃条件下的封闭体系中搅拌反应0.5~1h即可。在实际工业化生产中,本领域技术人员可根据实际情况及工业放大效应,选择适宜的搅拌反应时间,以达到三聚氰胺的充分成盐反应。
[0031]
其中,所述多元醇为季戊四醇、双季戊四醇和三季戊四醇中的至少一种,为统一实验设计及测试标准,进一步优选为季戊四醇,下述测试及分析结果均为采用季戊四醇的技
术方案。
[0032]
通常而言,在不添加其他组分或助剂的情况下,步骤(2)制备所得混合溶液应为黄色澄清透明溶液,步骤(3)制备所得单分子膨胀型阻燃剂mppr应为白色块状固体。
[0033]
通常而言,步骤(3)所述将步骤(2)所得混合溶液冷却后进行干燥至恒重,通常可选择静置冷却的方式,本领域技术人员也可根据现有技术或常规工业冷却技术进行冷却;其中所述干燥可选择鼓风干燥机进行干燥,例如放置在鼓风干燥机中采用90℃热风温度干燥至产品恒重,本领域技术人员也可根据现有技术或常规工业干燥技术进行干燥。
[0034]
上述单分子膨胀型阻燃剂mppr的制备方法中合成原理如下:
[0035]
植酸(pa)中含有六个磷酸基团,加热时其p
‑
oh结构可与季戊四醇(per)中的
‑
ch2‑
oh基团在水溶液中形成分子间氢键,从而形成pa
…
per氢键复合物,所得pa
…
per氢键复合物中间体可在加热条件下与三聚氰胺(me)发生成盐反应,从而制备出目标产物——具有单分子结构特点的氮
‑
磷膨胀型阻燃剂mppr,具体反应过程及机理可用说明书附图1表示。
[0036]
值得说明的是,本发明中所述植酸、季戊四醇、三聚氰胺分别作为酸源、炭源、气源的摩尔比为1:(2.4~2.6):(1.9~2.1),是通过本发明的发明人经实验对照并基于实验事实获得,在该配比下通过对所得单分子膨胀型阻燃剂mppr的热失重分析,表现出了良好的热稳定性,满足pp阻燃改性及成型加工要求。并且在该配比下,单分子膨胀型阻燃剂mppr中具有相较最优的磷含量及碳含量,具有极佳的综合阻燃性能。
[0037]
注意的是,本发明中限定了所用植酸、季戊四醇、三聚氰胺摩尔比为1:(2.4~2.6):(1.9~2.1)为主要保护范围,主要是基于本发明的发明人经大量实验及研究对比后提出的最优配比,本发明所要求保护的配比范围是在该最优配比上进行了适当的保护范围扩展,确保扩展后的配比范围与该最优配比的技术原理相同,并在技术效果上近乎一致。
[0038]
为了更好地说明本发明,并提供一种最优的技术方案,所述单分子膨胀型阻燃剂mppr,其原料组分包括植酸、季戊四醇、三聚氰胺,且植酸、季戊四醇、三聚氰胺摩尔比为1:2.5:2。
[0039]
通常地,其中所述聚丙烯选择可为市面上常规的聚丙烯原料。
[0040]
通常地,所述玻璃纤维(gf)为本技术领域中现有技术所使用的针对pp增强的玻璃纤维选择,本领域技术人员可根据现有技术的记载或报道选择市面上适宜的玻璃纤维种类或自行制备。为更方便进行制备加工处理,优选为短切玻璃纤维。
[0041]
通常地,所述马来酸酐接枝聚丙烯(mapp)为本技术领域中现有技术所使用的增容改性剂,本领域技术人员可根据现有技术的记载或报道选择市面上适宜用于增容改性具有合适接枝率的马来酸酐接枝聚丙烯。
[0042]
通常而言,本领域技术人员可根据上述mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料的配比,根据实际需求与现有技术选择适宜的共混加工成型方式,例如包括双螺杆挤出成型、热压成型、硫化成型等现有技术中记载的适于聚丙烯/填料体系共混成型制备工艺手段,从而制备得到mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料。
[0043]
为了更好地说明本发明,并提供一种优选的技术方案,上述mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料的制备方法,主要包括以下步骤:
[0044]
①
将主要包括单分子膨胀型阻燃剂mppr、聚丙烯、八乙烯基低聚倍半硅氧烷、玻璃纤维及马来酸酐接枝聚丙烯的组分原料混合均匀后作为混合料,通过双螺杆挤出机熔融挤
出造粒,收集所得复合材料粒料;其中,双螺杆挤出机的温度设置为160~200℃,挤出速度为100~150r/min;
[0045]
②
将步骤
①
所得复合材料粒料通过平板硫化机热压成型处理,即得mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料;其中,平板硫化机的热压温度为185℃~195℃,压力为10~15mpa。
[0046]
其中,上述制备方法中单分子膨胀型阻燃剂mppr、聚丙烯、八乙烯基低聚倍半硅氧烷、玻璃纤维及马来酸酐接枝聚丙烯,以重量份数计,为下述配比:
[0047][0048]
且总重量份数为100份。
[0049]
按照上述优选的技术方案制备所得mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料,垂直燃烧的性能达ul94 v
‑
0等级,极限氧指数达27.7~28.7%,其机械性能达到拉伸强度82.4mpa、缺口冲击强度13.89kj/m2和弯曲强度77.5mpa。
[0050]
本发明的主要发明点在于,本发明的发明人通过大量研究及实验数据研究证实了,当采用其自主研发的单分子膨胀型阻燃剂mppr添加至聚丙烯基体中后,虽然获得了综合阻燃性能极佳的阻燃聚丙烯复合材料,但pp材料的力学性能出现了不同程度的下降,其中阻燃样条(25mppr 75pp)的力学性能损失最大,其拉伸强度和缺口冲击强度分别下降至27.4mpa和2.42kj/m2。纯pp的力学性能其拉伸强度为32.0mpa,缺口冲击强度为3.15kj/m2。
[0051]
多面体低聚倍半硅氧烷(poss)是一种具有笼形刚性结构的有机
‑
无机纳米杂化材料,其粒径通常在1~3nm之间。现有研究表明,poss的热稳定性优良,已成功用于制备阻燃高分子材料。poss的通式为(rsio
1.5
)n,其中r为有机基团,例如乙烯基、氨基、苯基等。通过采用接枝、交联、熔融共混、共聚等方法可将poss分子和结构引入各种热塑性或热固性聚合物中,提高其力学性能、热稳定性和阻燃性能。在高温条件下,poss可被氧化成二氧化硅(sio2)并迁移至基体表面,防止形成的炭层热氧化和可燃气体释放,从而提高材料的热稳定性和阻燃性能,发挥其阻燃作用。八乙烯基倍半硅氧烷(ov
‑
poss)是poss的一种,其r基团为八乙烯基基团。
[0052]
现有技术中,通常认为八乙烯基倍半硅氧烷(ov
‑
poss)的加入量不宜过多,当超过22wt%时,poss纳米粒子发生了明显的团聚现象,而团聚的poss颗粒不能很好地分散在聚丙烯基体中。基于现有文献记载,将八乙烯基倍半硅氧烷(ov
‑
poss)与阻燃剂的复配使用,其添加量通常遵守等质量替换的原则(即1wt%的poss取代1wt%阻燃剂)。
[0053]
本发明的发明人发现将八乙烯基倍半硅氧烷(ov
‑
poss)引入本发明所提供的技术方案中,有利于改善mppr阻燃剂颗粒在pp基体中的分散性和界面相容性,使阻燃剂颗粒均匀地分散于pp基体树脂中。如说明书附图10所示,可以看出,当poss粒子与mppr阻燃剂颗粒混合时,在mppr颗粒表面包覆有许多poss纳米粒子,从而可实现对mppr阻燃剂颗粒表面改性,以此改善mppr与pp基体树脂的相容性及其分散性。总结起来,poss纳米粒子主要通过以
下方式起增容作用:一方面,poss纳米粒子本身与pp基体有很好的相容性;另一方面,poss纳米粒子比表面积大,其纳米尺寸效应引起的静电吸附作用易被吸附于阻燃剂颗粒表面,同时poss纳米粒子结构中的氧原子与mppr中氨基基团的氢原子可发生氢键相互作用。通过以上分析,可以合理地认为,引入的poss纳米粒子在体系中实际上起到小分子增容剂的作用。因此,经poss粒子包覆的单分子膨胀型阻燃剂mppr颗粒与pp基体之间的相容性显著改善,同时包覆有poss的单分子膨胀型阻燃剂mppr粒子在pp基体中的分散性也得到改善,从而有利于提高pp/mppr/poss阻燃复合材料的阻燃性能和力学性能。
[0054]
但是,经过进一步的研究,基于实验事实,通过八乙烯基倍半硅氧烷(ov
‑
poss)引入增容改性,其仅能够将制备所得阻燃聚丙烯复合材料的拉伸强度达32.3mpa,缺口冲击强度达2.92kj/m2。可明显看出,其机械性能更接近于纯pp,但是与纯pp相比,阻燃聚丙烯复合材料的力学性能仍呈现恶化趋势,不能满足其在需要满足高强度高模量应用场合(比如家用电器,汽车外壳等)的要求。
[0055]
于是,为了更进一步增强所得阻燃聚丙烯复合材料的机械性能,同时又需要保持其优秀的综合阻燃性能,本发明的发明人通过采用自主研发的玻璃纤维和马来酸酐接枝聚丙烯的复配体系,大幅提高了所得阻燃聚丙烯复合材料的机械性能,同时保障了原有的综合阻燃性能。
[0056]
其中,值得重点说明的是,若仅通过单独添加玻璃纤维的作为填料的方式,玻璃纤维所具有的“烛芯效应”可能会损坏最终产品的阻燃性能,此外,由于聚丙烯与玻璃纤维是热力学不相容体系,其差的界面结合作用也会限制基体的力学性能提升,因此在此基础上,本发明的发明人通过玻璃纤维和马来酸酐接枝聚丙烯的复配结合体系进行增容改性,大幅改善了阻燃聚丙烯复合材料的界面结合,并抑制了其“烛芯效应”缺陷。
[0057]
需要注意的是,当玻璃纤维的占比低于20wt%时,通过对照实验结果证实其样品阻燃性出现了明显的下降,推测是与单分子膨胀型阻燃剂mppr含量较低所导致;而适当增加玻璃纤维有利于阻燃聚丙烯复合材料的阻燃性能,当玻璃纤维含量为20wt%~30wt%时,均可达到垂直燃烧ul94 v
‑
0级别,并且材料的phrr值和thr值亦明显降低;但是当玻璃纤维的含量明显超过30wt%时,因部分未被残炭阻断的玻纤“烛芯效应”会恶化阻燃玻纤增强pp复合材料的阻燃性能,并通过对照实验确认了,继续增加玻纤负载量会导致材料力学性能下降。
[0058]
mppr主要发挥气相和凝聚相阻燃作用;玻纤一方面起着稀释聚合物浓度作用,另一方面主要通过与膨胀炭层形成交联网络结构提高炭层强度,避免材料产生熔滴,提高材料阻燃性能;而poss主要发挥凝聚相阻燃作用,由其分解产生的sio2迁移至炭层表面,与炭渣一起包覆于玻纤表面,从而中断玻纤的热传导机制,抑制其“烛芯效应”。上述阻燃体系中各组分作用相互促进,共同提高阻燃pp材料的阻燃性能。
[0059]
同时,可直接通过肉眼观察到,所得mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料作为阻燃样条在点燃后都产生了膨胀炭层,这是mppr中的酸源(pa)部分受热分解产生无机酸(偏磷酸),无机酸催化碳源(per)部分发生成炭反应,同时气源部分分解产生的不燃性气体(nh3、n2等)使炭层发生膨胀引起。生成的膨胀炭层能有效隔绝pp基体和外界热量和氧气的传递,阻止底层基体的进一步燃烧,从而实现阻燃目的。
[0060]
其中,步骤
①
中所述将主要包括单分子膨胀型阻燃剂mppr、聚丙烯、八乙烯基低聚
倍半硅氧烷、玻璃纤维及马来酸酐接枝聚丙烯的组分原料混合均匀后作为混合料,可选择在添加前,将混合料与80~90℃恒温环境下鼓风干燥至恒重。
[0061]
值得说明的是,本发明的发明人通过实际的对照实验发现,当选择添加1~2wt%的八乙烯基低聚倍半硅氧烷,能够起到减少4~5wt%单分子膨胀型阻燃剂mppr的添加量,并具有最佳的阻燃综合性能,并且可以明显提高pp/mppr阻燃体系凝聚相的成炭能力。
[0062]
但是,需要重点说明的是,继续提高协效阻燃剂poss的取代量(大于2wt%)会降低体系的残炭率,从而劣化材料的阻燃性能。这主要有下述两方面原因:一方面,poss本身的成炭能力比mppr差,poss含量增加意味着发挥主要阻燃作用的主阻燃剂mppr含量相应减少,因此mppr的相对减少会影响材料的阻燃性能,导致阻燃材料的残炭率降低;另一方面,添加较多的poss纳米颗粒容易团聚,从而影响主阻燃剂mppr在基体树脂中的分散以及poss自身参与pp大分子链的成炭反应,降低阻燃pp复合材料的成炭能力和残炭率。同时,降低协效阻燃剂poss的取代量(小于1wt%)会降低体系的机械性能,其原因同样为阻燃剂mppr与pp基体的界面相容性较差。
[0063]
通常地,除所述组分外,本发明还可添加其它现有技术公知的加工助剂,例如颜料、抗氧剂等。但前提是,这些加工助剂对本发明的目的实现以及对本发明优良效果的取得不得造成不利影响。
[0064]
本发明技术方案具有以下有益效果:
[0065]
(一)本发明提供了一种单分子膨胀型阻燃剂mppr,将其与聚丙烯混合制备mppr阻燃聚丙烯复合材料,通过垂直燃烧、极限氧指数和锥形量热测试结果表明,加入单分子膨胀型阻燃剂mppr显著提高了聚丙烯的阻燃性能。此外,通过限定单分子膨胀型阻燃剂mppr的制备方法,通过其反应机理大幅缩短制备所需时间,相较现有的单分子膨胀型阻燃剂制备工艺极大地降低了工业制备所需用时。
[0066]
(二)本发明采用热重分析和对凝聚相炭层的sem分析证明了添加有单分子膨胀型阻燃剂mppr的阻燃聚丙烯复合材料的综合阻燃性能。结果表明,mppr兼具气相和凝聚相阻燃作用。对于气相阻燃,mppr受热分解产生不燃性气体,稀释了可燃气体浓度,抑制材料的燃烧。对于凝聚相阻燃,mppr酸源通过受热分解产生的磷酸催化部分炭源发生成炭反应及促进聚合物基体的炭化,同时受热分解产生的气体使炭层发生膨胀,形成膨胀炭层,隔绝外界热氧的传递,阻止底层基体的进一步燃烧,从而实现阻燃作用。
[0067]
(三)本发明通过实验数据结果分析说明了mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料更优的综合阻燃效果和机械性能。其中,mppr主要发挥气相和凝聚相阻燃作用;玻纤一方面起着稀释聚合物浓度作用,另一方面主要通过与膨胀炭层形成交联网络结构提高炭层强度,避免材料产生熔滴,提高材料阻燃性能;而poss主要发挥凝聚相阻燃作用,由其分解产生的sio2迁移至炭层表面,与炭渣一起包覆于玻纤表面,从而中断玻纤的热传导机制,抑制其“烛芯效应”。
[0068]
(四)本发明所提供的mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料的制备方法,工艺简单,流程短,生产过程无三废污染,可为制备综合性能优异的膨胀阻燃pp复合材料提供新方法新技术。
附图说明
[0069]
图1为本发明单分子膨胀型阻燃剂mppr制备方法的具体反应过程及机理示意图。
[0070]
图2为本发明合成例1制备所得单分子膨胀型阻燃剂mppr的红外光谱分析图。可以看出,在pa的红外谱图中,pa对应的特征吸收峰分别为3410cm
‑1(p
‑
oh),1061cm
‑1(p=o)和859cm
‑1(p
‑
o
‑
c中的p
‑
o);在per谱图中,3326cm
‑1是c
‑
oh特征吸收峰。与pa和per相比,中间体pa
…
per氢键复合物的
‑
oh吸收峰的位置为3332cm
‑1,处于二者之间。相较于pa,pa
…
per的
‑
oh吸收峰向低波数方向移动,发生了红移。产生该变化的原因是per的加入破坏了pa与pa之间的分子间氢键,同时在pa与per之间形成了新的分子间氢键pa
…
per,新的分子间氢键相互作用力比pa自身氢键作用更强。此外,中间体pa
…
per的p
‑
o吸收峰从859cm
‑1向高波数方向移动至872cm
‑1,这是由于在氢键作用下,p和o之间的相互作用减弱所引起的。与中间体pa
…
per相比,mppr的红外谱图中出现了一系列新的特征吸收峰。其中,特征吸收峰1673cm
‑1、1499cm
‑1和1403cm
‑1来源于me的三嗪环,3364cm
‑1为
‑
nh2的伸缩振动吸收,3143cm
‑1为nh
+
的振动吸收,1174cm
‑1则对应于po2‑
的特征吸收,说明pa已与me在水溶液中发生了成盐反应,生成了
‑
po
‑
·
hn+≤官能基团。以上红外分析结果证明了图中发生的相关反应,同时也表明成功合成了单分子膨胀型阻燃剂mppr。
[0071]
图3为本发明合成例1制备所得单分子膨胀型阻燃剂mppr的tg和dtg曲线图。从图中可以看出,mppr只有一个主分解峰,其初始热分解温度为238℃,最大热分解速率约为0.28%/℃,而pp的加工温度一般在180~220℃之间。此外,mppr在700℃的残炭率高达41.9%,其高残炭率也有利于高效发挥阻燃剂的阻燃作用。因此,制备得到的mppr具有良好的热稳定性,可以满足pp阻燃改性及成型加工要求。
[0072]
图4为本发明应用例1~3、应用对比例1制备所得样品在垂直燃烧测试中的照片,图中(a)为应用对比例1制备所得样品,(b)为应用例3制备所得样品,(c)为应用例2制备所得样品,(d)为应用例1制备所得样品。可以看出,pp/mppr(82/18,w/w)样品的阻燃性能较差,点火后一直燃烧至夹具,不能自熄,因此不能通过垂直燃烧等级测试;添加20wt%mppr后,复合材料第二次点火后只有通过熔滴滴落带走部分热量才能使燃烧火焰熄灭,故只能达到ul94 v
‑
2(3.2mm)等级;添加22wt%mppr时,pp阻燃材料燃烧时无熔滴产生,但两次点火自熄时间之和超过10s,根据ul94标准可达到ul94 v
‑
1级别;进一步增加阻燃剂含量至25wt%后,pp/mppr复合材料可达到ul94 v
‑
0等级,阻燃性能明显提升。同时,还可以看到,所有的pp/mppr阻燃样条在点燃后都产生了膨胀炭层,这是mppr中的酸源(pa)部分受热分解产生无机酸(偏磷酸),无机酸催化碳源(per)部分发生成炭反应,同时气源部分分解产生的不然性气体(nh3、n2等)使炭层发生膨胀引起。生成的膨胀炭层能有效隔绝pp基体和外界热量和氧气的传递,阻止底层基体的进一步燃烧,从而实现阻燃目的。
[0073]
图5为本发明应用例1制备所得样品在锥形量热测试结果曲线图。从(a)、(b)中可以看出,纯pp的点燃时间为40s,点燃后在较短时间(40~300s)内释放出大量热量,其phrr值和thr值分别达到1463kw/m2和155mj/m2。与纯pp相比,应用例1制备所得样品(25mppr75pp)的点燃时间提前到26s,这归因于阻燃剂促进了pp的提前降解。此外,阻燃pp材料的phrr值降低至189kw/m2,thr降低至139mj/m2,阻燃剂mppr的加入显著降低了pp复合材料的hrr和thr,证明其能明显提升pp阻燃性能。从(c)、(d)中可以看出,相对于纯pp,应用例1制备所得样品(25mppr75pp)的spr和tsp曲线呈明显下降趋势,其烟释放速率和总烟雾
释放量分别从纯pp样品的0.155m2/s和20.3m2下降至0.038m2/s和16.4m2,这表明mppr加入对烟气释放有抑制作用。
[0074]
图6为本发明应用例1制备所得样品的质量损失与时间的关系曲线图。很明显看出,加入mppr后,阻燃pp复合材料的平均质量损失率有所下降,其锥形量热测试后的残炭率高达25.1%,说明mppr有促进成炭的作用。
[0075]
图7为本发明应用例1制备所得样品(b)与纯pp(a)在锥形量热仪测试后的残炭数码照片。可见,纯pp在燃烧之后没有残留物,但应用例1制备所得样品在燃烧之后仍有大量残炭形成,且形成的炭层膨胀程度较高,说明mppr在燃烧过程中通过释放惰性气体而发挥其气相阻燃作用。
[0076]
图8为本发明应用例1、应用例3及应用对比例1制备所得样品的扫描电镜(sem)图,其中(a)、(b)为应用对比例1的样品,(c)、(d)为应用例3的样品,(e)、(f)为应用例1的样品。从sem图中可以看出,随着阻燃剂mppr含量的增加,炭层致密性和膨胀程度逐渐增加。当mppr添加量增至25wt%时,燃烧后形成的炭层连续且致密,炭层质量大幅改善。一般而言,炭层可以利用其对外界氧气和基体树脂分解产生的可燃性气体阻隔作用影响材料的阻燃性能。因此,pp/mppr阻燃材料在燃烧过程中形成的炭层可较好发挥其凝聚相阻燃作用。因此可知,mppr在燃烧过程中可同时发挥气相和凝聚相阻燃作用,对提升pp材料阻燃性能效果显著。
[0077]
图9为本发明应用例1制备所得样品在500℃下形成的残炭的c
1s
和p
2p
的xps光谱图。xps测试结果证实在燃烧过程中形成了一些具有稳定化学结构的产物。在c1s光谱中,284.7ev处的峰归属于c
‑
c或c=c键,而在285.7ev和288.6ev附近的能谱峰分别归因于阻燃pp材料降解产物中的c
‑
o(和/或c
‑
n)和c=o键。p
2p
光谱中出现了两个不同的能谱峰,其中134.8ev附近的能谱峰归属于o=p
‑
o
‑
c基团,134.1ev附近的能谱峰归因于焦磷酸盐和/或多聚磷酸盐,说明阻燃剂中的p元素在pp/mppr复合材料降解过程中参与了凝聚相成炭反应。
[0078]
图10为本发明应用例4、应用对比例2、应用对比例3制备所得样品脆断面的扫描电镜(sem)图。可以看出,应用对比例2样品(a和b)脆断表面较为平整,未出现孔洞等缺陷,这是由于poss粒子尺寸较小,与基体相容性较好,但同时也注意到,由于poss加入量(22wt%)过多,poss纳米粒子发生了明显团聚现象,团聚的poss颗粒不能很好地分散在聚丙烯基体中。应用对比例3样品(c和d)脆断表面粗糙,存在明显的团聚阻燃剂颗粒和较大孔洞(脆断时阻燃剂颗粒与pp基体分离而形成),说明阻燃剂颗粒在pp基体中相容性和分散性较差;应用例4样品(e和f)断面较平整,样品表面暴露的阻燃剂颗粒较少,缺陷导致的孔洞直径明显减小。这是因为poss粒子的存在有利于改善mppr阻燃剂颗粒在pp基体中的分散性和界面相容性,使阻燃剂颗粒均匀地分散于pp基体树脂中。
[0079]
图11为本发明应用例4、应用对比例3制备所得样品及纯pp(pp0)的锥形量热测试结果曲线图。从(a)可以看出,纯pp(pp0)燃烧迅速而剧烈,其热释放速率(hrr)曲线显示出很强的单峰,说明pp在较短时间内(30~350s)释放大量热量。纯pp(pp0)的峰值hrr(phrr)达到1025kw/m2。当添加22wt%的mppr填料时,应用对比例3样品(pp2)的phrr值大大降低至315kw/m2。应用例4样品(pp3)的phrr值进一步降至252kw/m2,达到phrr的时间(t
‑
phrr
)延迟到200s。同时,还注意到在200s后pp3的hrr呈持续降低趋势,这表明相应pp/mppr/poss体系
中的凝聚相成炭反应仍在持续进行,所形成的凝聚相炭层质量在不断改善。由此可见,当少量mppr阻燃剂被poss粒子取代时并不会降低材料的阻燃性能,反而会明显提高材料阻燃性能。
[0080]
图12为本发明应用例4、应用对比例3制备所得样品及纯pp(pp0)的质量损失与时间之间的关系曲线。显然纯pp的质量随时间迅速减少,几乎没有残炭剩余。在pp中加入mppr或mppr/poss复合阻燃剂后,阻燃pp复合材料在燃烧过程中的质量下降速率得到了明显抑制,残炭的质量保留率显著增加。此外,通过比较pp2和pp3的质量损失曲线,发现后者在不同时间的炭残留质量均明显高于前者的炭残留质量,所得pp2和pp3的最终残炭率分别为10.8%和18.0%。显然,mppr/poss协效阻燃体系在促进炭层的形成和降低放热速率方面均优于单独使用mppr阻燃剂,能更好地减缓pp基体材料的燃烧。
[0081]
图13为本发明应用例4、应用对比例3制备所得样品在锥形量热试验后阻燃pp材料炭层的扫描电镜(sem)图。可以看出,pp2样品(a和b)的炭层虽然膨胀程度较高,但存在较多孔洞,燃烧时不能有效隔绝基体树脂与外界热氧的接触。相反,应用例4样品(pp3)的炭层质量显著提高(c和d),形成了连续致密的炭层。此外,如果仔细观察阻燃样品pp3的炭层sem照片(d),会发现样品燃烧后炭层表面明显覆盖有许多细小的白色颗粒。为了确定这些白色颗粒物质的成分,采用eds对pp2和pp3炭层进行了对比分析,结果如图所示,可见,pp2样品的残炭中由c、o和p三种元素组成,而pp3样品的残炭表面还含有si元素。因此,可以合理地认为覆盖于pp3炭层表面的白色颗粒物质是由样品中引入的poss分解形成的sio2粒子。这些sio2颗粒可以有效地增强炭层强度并防止炭层氧化降解,从而阻止燃烧过程中可燃气体和热量的传递,表现出poss优异的协效阻燃作用。
[0082]
图14为本发明应用例6、8,应用对比例7、8所得样品在氮气气氛下的热重分析曲线图。
[0083]
图15为本发明应用例6、8,应用对比例7、8所得样品的微型燃烧量热(mcc)曲线图。从图中可以看出,玻纤含量的增加显著降低了材料的最大热释放速率(phrr),总热释放量(thr)等值。例如玻纤添加量为10wt%时,材料的最大热释放速率为394.9w/g,总热释放量为30.3kj/g;玻纤含量为30wt%时,gfpp3材料的phrr降低至336.1w/g,thr降低至22.3kj/g,与gfpp5相比分别降低了14.9%和26.4%;当玻纤含量增至40wt%,gfpp7的phrr和thr分别显著降至243.2w/g和16.3kj/g。从tga和燃烧测试结果已知,将gf添加到gfpp复合材料中可以改善协效阻燃剂mppr/poss在燃烧过程中形成的炭层的强度,更有效地抑制可燃挥发物的释放。同时,分散在pp基体中的gf降低了聚丙烯所占比重,使可燃性高聚物浓度下降,一定程度上减少了燃烧物浓度,发挥出其稀释作用。因此,玻纤的加入可以降低pp材料的热释放情况,并在一定程度上提高其阻燃性。同时,还可注意到,虽然gfpp7复合材料的phrr和thr均低于gfpp3,但前者阻燃性能却更差,这可能是由于玻纤含量更多的前者基体树脂pp含量更少(导致phrr和thr更低),且部分未被炭层覆盖的玻纤“烛芯效应”所致。
[0084]
图16为本发明应用例6,应用对比例7、8所得样品的在垂直燃烧测试后的残炭电镜图。其中,应用对比例7的样品残炭电镜图为(a)、(a’),应用例6的样品残炭电镜图为(b)、(b’),应用对比例8的样品残炭电镜图为(c)、(c’)。(a)、(b)、(c)为炭层外表形貌,(a’)、(b’)、(c’)为炭层内部形貌。可以看出,玻纤含量较少时(gfpp5和gfpp3),mppr分解产生的膨胀炭层可较完整地覆盖于玻纤表面,其中玻纤通过与膨胀炭层形成交联网络结构增强炭
层强度,限制了热量和可燃性气体向被阻燃pp基体树脂内部扩散,达到提高阻燃性能目的。当玻纤含量增至40wt%时,炭层表面出现较大孔洞,显然易导致基底pp树脂的燃烧,劣化材料的阻燃性能。此外,还可以看出,玻纤含量低于40wt%时,在炭层内部包覆于玻纤表面的炭渣和由poss原位分解产生的sio2颗粒较多,抑制了玻纤的热传导并切断了其传播路径,从而减弱了其“烛芯效应”,但当玻纤含量增至40wt%时,玻纤表面覆盖的炭残留物减少,导致玻纤“烛芯效应”增强使材料阻燃性能下降。
[0085]
图17为本发明应用例6、7,应用对比例5、6所得样品的脆断面形貌电镜图。其中,gfpp1(a),gfpp2(b),gfpp3(c),gfpp4(d)。可以看出,不加mapp时,材料脆断面存在大量玻纤拔出的孔隙(gfpp1),加入mapp后,玻纤拔出造成的孔隙明显减少,mapp的加入明显改善了玻纤与pp基体之间的界面结合强度和相容性,这也是加入mapp后材料力学性能改善的直接证据和原因。
具体实施方式
[0086]
下面通过实施例并结合附图对本发明作进一步说明。值得指出的是,给出的实施例不能理解为对本发明保护范围的限制,该领域的技术人员根据本发明的内容对本发明作出的一些非本质的改进和调整仍应属于本发明保护范围。
[0087]
下述合成例及应用例、应用对比例所采用的实验原料和试剂:
[0088]
聚丙烯(pp):t30s,中国石化股份有限公司茂名分公司;
[0089]
植酸(pa):阿拉丁生化科技股份有限公司;
[0090]
三聚氰胺(me):工业纯,纯度99.8%,成都玉龙化工股份有限公司;
[0091]
季戊四醇(per):纯度98.0%,天津市光复精细化工研究所;
[0092]
双季戊四醇(dper):纯度95.0%,安徽金禾实业股份有限公司;
[0093]
三季戊四醇(tper):纯度95.0%,武汉富鑫远科技有限公司;
[0094]
去离子水:市售;
[0095]
八乙烯基低聚硅倍半氧烷(ov
‑
poss):郑州阿尔法化工有限公司;
[0096]
短切玻璃纤维(gf):508a,巨石集团有限公司;
[0097]
马来酸酐接枝聚丙烯(mapp):接枝率1.2%,广东省东莞市康锦新材料
[0098]
科技有限公司;
[0099]
抗氧剂1010:纯度99%,东莞市康锦新材料科技有限公司。
[0100]
下述合成例及应用例、应用对比例所采用的主要实验设备:
[0101]
电热恒温鼓风干燥箱:dhg
‑
9245a型,上海一恒科技仪器有限公司;
[0102]
转矩流变仪:rm
‑
200c型,哈尔滨哈普电气技术有限责任公司;
[0103]
平板硫化压机:yj63型,成都瑞琪创鸿制造有限公司;
[0104]
双螺杆挤出机:tssj
‑
25型,中蓝晨光化工研究院科强化工装备公司;
[0105]
傅立叶变换红外光谱仪(ft
‑
ir):nicilet6700型,赛默飞世尔科技公司;
[0106]
极限氧指数测试仪:jf
‑
3型,南京江宁分析仪器有限公司;
[0107]
垂直(水平)燃烧一体测试仪:lzb
‑
3wb型,东莞市金特仪器有限公司;
[0108]
锥形量热仪(cone):ftt0007型,英国ftt公司;
[0109]
微型燃烧量热仪(mcc):faa型,英国燃烧测试技术公司;
[0110]
x射线光电子能谱仪(xps):axis supra型,英国kratos公司;
[0111]
摆锤冲击试验机:pit
‑
501j型,深圳万测试验设备有限公司;
[0112]
缺口型制样机:4302instron型,深圳市新三思材料检测有限公司;
[0113]
万能试验机:instron 5967型,因斯特朗公司;
[0114]
扫描电子显微镜(sem):xsam800型,美国fei公司;
[0115]
热重分析仪(tga):q50型,美国ta公司;
[0116]
差示扫描量热仪(dsc):q20型,美国ta公司;
[0117]
拉曼光谱分析仪:ra802型,英国renishaw公司;
[0118]
熔体流动速率试验机:zrz1442型,美特斯工业系统(中国)有限公司。
[0119]
下述合成例及应用例、应用对比例所采用的测试和表征方法:
[0120]
傅立叶变换红外光谱(ft
‑
ir)分析:采用kbr压片法制样,使用nicolet 20sxb红外ft
‑
ir光谱仪在400至4000cm
‑1之间扫描和记录傅立叶红外光谱图。
[0121]
热重(tga)分析:采用q50型热重分析仪(ta公司,usa),在氮气或空气气氛下,以10℃/min的升温速率在50至700℃的温度范围对样品进行热重分析(tga),气体流速为50ml/min,样品质量约为5mg。
[0122]
差示扫描量热(dsc)分析:采用q
‑
20型差示扫描量热仪(ta公司,usa)记录样品的dsc曲线。样品质量约7mg,并在50ml/min的氮气流量以10℃/min的加热速率在0至200℃之间进行测量。
[0123]
微型燃烧量热(mcc)测试:取约4~5mg的样品,在80ml/min的氮气气氛条件下以1℃/s的加热速率于faa型微型燃烧量热仪(英国燃烧测试技术公司)上测量样品的热释放情况,其中测试温度范围为80℃至750℃。
[0124]
极限氧指数(loi)测试:根据astm d2863
‑
97标准,在jf
‑
3型氧指数测试仪上进行loi测试,样品尺寸为100mm
×
6.5mm
×
3.0mm。
[0125]
垂直燃烧ul94测试:根据astm d 3801测试标准在垂直(水平)燃烧一体测试仪上进行垂直燃烧性能测试,样品尺寸为127mm
×
12.7mm
×
3.2mm。
[0126]
合成例1
[0127]
本合成例单分子膨胀型阻燃剂mppr的制备方法,包括以下步骤:
[0128]
(1)将0.05mol植酸与200ml去离子水配置为植酸溶液;
[0129]
(2)将季戊四醇加入步骤(1)所得的植酸溶液,于95~100℃条件下搅拌反应1h;再加入三聚氰胺,继续于95~100℃条件下搅拌反应0.5h,即得黄色澄清透明的混合溶液;
[0130]
(3)将步骤(2)所得混合溶液冷却后进行干燥至恒重,即得白色块状的单分子膨胀型阻燃剂mppr。
[0131]
其中,上述制备方法中所用植酸、季戊四醇、三聚氰胺摩尔比为1:2.5:2。
[0132]
合成例2
[0133]
本合成例单分子膨胀型阻燃剂mppr的制备方法,包括以下步骤:
[0134]
(1)将0.1mol植酸与600ml去离子水配置为植酸溶液;
[0135]
(2)将双季戊四醇加入步骤(1)所得的植酸溶液,于95~100℃条件下搅拌反应1.5h;再加入三聚氰胺,继续于95~100℃条件下搅拌反应1h,即得黄色澄清透明的混合溶液;
[0136]
(3)将步骤(2)所得混合溶液冷却后进行干燥至恒重,即得白色块状的单分子膨胀型阻燃剂mppr。
[0137]
其中,上述制备方法中所用植酸、双季戊四醇、三聚氰胺摩尔比为1:2.4:2.1。
[0138]
合成例3
[0139]
本合成例单分子膨胀型阻燃剂mppr的制备方法,包括以下步骤:
[0140]
(1)将0.15mol植酸与1000ml去离子水配置为植酸溶液;
[0141]
(2)将三季戊四醇加入步骤(1)所得的植酸溶液,于95~100℃条件下搅拌反应2h;再加入三聚氰胺,继续于95~100℃条件下搅拌反应1h,即得黄色澄清透明的混合溶液;
[0142]
(3)将步骤(2)所得混合溶液冷却后进行干燥至恒重,即得白色块状的单分子膨胀型阻燃剂mppr。
[0143]
其中,上述制备方法中所用植酸、三季戊四醇、三聚氰胺摩尔比为1:2.6:1.9。
[0144]
应用例1~3、应用对比例1
[0145]
本应用例1~3及应用对比例1是利用合成例1制备所得单分子膨胀型阻燃剂mppr与聚丙烯混合制备mppr阻燃聚丙烯复合材料,即一种mppr阻燃聚丙烯复合材料的制备方法,主要包括以下步骤:
[0146]
①
将主要包括单分子膨胀型阻燃剂mppr与聚丙烯的组分原料混合均匀后作为混合料,添加至转矩流变仪熔融共混,密炼处理5~10min,收集所得密炼混合物;其中,转矩流变仪的螺杆转速为40~50rpm;
[0147]
②
将步骤
①
所得密炼混合物通过平板硫化机热压成型处理,即得mppr阻燃聚丙烯复合材料;其中,平板硫化机的热压温度为190℃。
[0148]
其中,上述制备方法中聚丙烯与单分子膨胀型阻燃剂mppr,以重量份数计,应用例1~3、应用对比例1为下述配比:
[0149] 聚丙烯单分子膨胀型阻燃剂mppr应用例17525应用例27822应用例38020应用对比例18218
[0150]
将上述应用例1~3、应用对比例1与纯pp进行垂直燃烧ul94测试和极限氧指数测试,结果如下表:
[0151][0152]
将上述应用例1与纯pp进行锥形量热测试,结果如下表:
[0153][0154]
上表给出的ehc值反映了气相中挥发性气体的燃烧程度。可以看出,纯pp的ehc值为39.5mj/kg,而应用例1制备所得样品的ehc值降低至33.8mj/kg。显然,阻燃剂mppr的添加会明显降低pp材料的ehc和hrr值,表明添加的mppr在燃烧过程中主要在气相和冷凝相中发挥阻燃作用。一方面,mppr分解产生的不可燃气体(如co2和nh3)稀释了由pp降解产生的可燃气体浓度,从而在一定程度上降低了材料的燃烧程度。另一方面,由mppr中的酸源分解产生的无机酸催化碳源发生脱水反应并形成炭层,从而隔离热氧并防止pp基体进一步燃烧。
[0155]
将上述应用例1~3、应用对比例1与纯pp在氮气气氛和空气气氛下分别进行热重分析,结果如下表:
[0156]
[0157][0158]
明显看出,在n2气氛下,纯pp的初始分解温度(t
onset
)为407.1℃,t
max
(最大分解速率对应的温度)值为447.9℃,降解后残炭率为0。加入阻燃剂mppr后,所有阻燃pp样品的初始分解温度均低于纯pp,这一方面可能是由加入的阻燃剂mppr分解造成,另一方面可能是加入的mppr促进了pp的早期分解(240~410℃)。此后,随着降解的进行,在410℃以后,pp/mppr复合材料的降解速率低于纯聚丙烯材料,这是因为mppr在早期分解阶段促进了pp/mppr复合体系膨胀炭层的形成,抑制了pp基体的降解,因此,阻燃聚丙烯的热稳定性逐渐高于纯pp。在最后分解阶段,pp/mppr阻燃材料的最大分解速率对应的温度值和在700℃下的残炭率均高于纯pp,其中pp/mppr(75/25,w/w)复合材料的t
max
值457.7℃,最终残炭率高达11.4%,在所有阻燃材料中表现出最好的热稳定性。这是因为mppr改善了pp阻燃样品在高温下形成的残炭热稳定性,有利于提高其阻燃性能。
[0159]
在空气气氛下,pp和pp/mppr阻燃样品的热降解行为发生明显变化。纯pp的t
onset
值和700℃下的残炭率分别为242.7℃和0.1%。与纯pp相比,pp/mppr(75/25,w/w)样品的初始分解温度和t
max
值分别提高了5.3%和106%,最大热分解速率降低了约40%。上述分析结果说明,加入mppr后,所有阻燃pp样品的t
onset
值都明显高于纯pp,且材料的t
max
值随着mppr含量的增加而增大,说明阻燃剂的加入有利于提高pp/mppr阻燃复合材料在空气中的热稳定性,进而提高其阻燃性能。
[0160]
将上述应用例1~3、应用对比例1与纯pp进行差示扫描量热分析,结果如下表:
[0161]
可明显看出,mppr的加入改变了阻燃pp材料的熔融结晶行为,pp/mppr阻燃复合材料的初始结晶温度(t
onset
‑
c
)和结晶峰温度(t
c
)明显高于纯pp,表明mppr对pp结晶具有异相成核作用。此外,随mppr含量的增加,材料的熔融温度(tm)和结晶度(xc)呈增大趋势。这可能是因为mppr在结晶过程中扮演了成核剂的角色,增加mppr含量意味着其异相成核效应的
增强,这使得pp可以在较高温度下结晶,此时pp大分子链活动性较高,能够形成晶体完善性更高的结晶,从而有利于提高pp/mppr复合材料的结晶度,然而亦可能会导致材料冲击性能降低,这与后面力学性能测试结果一致。
[0162]
将上述应用例1~3、应用对比例1与纯pp进行力学性能测试,结果如下表:
[0163][0164]
可以看出,纯pp的力学性能最好,其拉伸强度为32.0mpa,缺口冲击强度为3.15kj/m2。加入阻燃剂后,pp材料的力学性能有不同程度的下降。
[0165]
结合ul94、loi、cone、tga、sem和xps的分析结果,mppr阻燃聚丙烯的阻燃机理总结如下:首先,mppr中的酸源(pa)部分受热分解产生磷酸,磷酸催化炭源(per)部分发生成炭反应,并加快了聚合物基体的炭化。同时,mppr受热分解产生不燃性气体nh3和co2等。这些不燃气体一方面稀释了可燃气体浓度,抑制材料的燃烧;另一方面,逸出的气体会使炭层发生膨胀形成膨胀炭层,隔绝外界氧气和热量的传递,阻止聚合物底层基体的进一步燃烧,从而实现阻燃效果。因此,阻燃剂mppr兼具气相和凝聚相阻燃作用。
[0166]
应用例4~5、应用对比例2~4
[0167]
本应用例4~5及应用对比例2~4是利用合成例1制备所得单分子膨胀型阻燃剂mppr与聚丙烯、八乙烯基低聚倍半硅氧烷混合制备mppr/poss复合协效无卤阻燃聚丙烯复合材料,即一种mppr/poss复合协效无卤阻燃聚丙烯复合材料的制备方法,主要包括以下步骤:
[0168]
①
将主要包括单分子膨胀型阻燃剂mppr、聚丙烯、八乙烯基低聚倍半硅氧烷的组分原料混合均匀后作为混合料,添加至密炼机中,密炼处理5~10min,收集所得密炼混合物;其中,密炼处理的温度为180~200℃,螺杆转速为40~50rpm;
[0169]
②
将步骤
①
所得密炼混合物冷却后破碎造粒,即得mppr/poss复合协效无卤阻燃聚丙烯复合材料。
[0170]
其中,上述制备方法中单分子膨胀型阻燃剂mppr、聚丙烯、八乙烯基低聚倍半硅氧烷(ov
‑
poss),以重量份数计,应用例4~5、应用对比例2~4为下述配比:
[0171] 聚丙烯单分子膨胀型阻燃剂mpprov
‑
poss应用对比例2(pp1)78022应用对比例3(pp2)78220应用例4(pp3)78211应用例5(pp4)78202应用对比例4(pp5)78184
[0172]
将上述应用例4~5、应用对比例2~4与纯pp(pp0)与纯ov
‑
poss在氮气气氛下进行热重分析,结果如下表:
[0173][0174]
可以看出,在氮气气氛下,纯pp在410℃至480℃之间迅速分解,其最大热分解速率所对应的温度(t
max
)出现在447.9℃。而所有加入阻燃剂的pp复合材料的初始分解温度(t
onset
)均低于纯pp,这归因于加入的阻燃剂的早期分解。同时,ov
‑
poss在氮气气氛下表现出一步降解过程(230~300℃),且其残炭率仅为2.6%。与纯pp和poss相比,pp/poss样品(pp1)的残炭率达2.9%,高于其理论计算值0.64%,说明poss参与了pp分子链的成炭反应。此外,在300~340℃之间,poss的加入还促进了pp的降解,说明poss粒子受热分解产生的无机sio2粒子参与了后续阻燃体系中的凝聚相炭层反应,这也将在后续的炭层sem和eds中得到进一步验证。pp/mppr和pp/mppr/poss样品的t
max
值比纯pp更高,这是因为阻燃剂mppr在初始阶段分解形成的膨胀炭层隔绝了内部基体材料和外部氧气的接触,在一定程度上抑制了pp基体的降解。当温度增至700℃时,纯pp完全分解且无任何剩余残炭,但含有mppr/poss复合物或mppr的阻燃pp复合材料的残炭率显著增加,尤其对相同负载量的阻燃体系,含1wt%poss的阻燃pp样品(pp3)在700℃时可达到最大残炭率(10.1%),高于不含poss的阻燃pp样品(pp2,残炭率9.0%)。这表明在阻燃pp复合材料中加入1wt%poss可以明显提高pp/mppr阻燃体系凝聚相的成炭能力。继续提高协效阻燃剂poss的取代量(2wt%或更高)会降低体系的残炭率,从而劣化材料的阻燃性能。这主要有下述两方面原因:一方面,poss本身的成炭能力比mppr差,poss含量增加意味着发挥主要阻燃作用的主阻燃剂mppr含量相应减少,因此mppr的相对减少会影响材料的阻燃性能,导致阻燃材料的残炭率降低;另一方面,添加较多的poss纳米颗粒容易团聚,从而影响主阻燃剂mppr在基体树脂中的分散以及poss自身参与pp大分子链的成炭反应,降低阻燃pp复合材料的成炭能力和残炭率。
[0175]
将上述应用例4~5、应用对比例2~4与纯pp(pp0)与纯ov
‑
poss在空气气氛下进行热重分析,结果如下表:
[0176][0177]
可以看出,阻燃pp样品在不同的载气气氛(空气与氮气)下具有不同的降解行为。在空气气氛下,纯pp的t
onset
值为259.3℃,t
max
为325.5℃,在700℃时残炭率为0.1%,表现出一步降解过程。poss粒子在空气气氛下则表现出两步降解过程(200~300℃和300~700℃),其t
onset
值和t
max
值分别为226.0℃和227.5℃。与氮气气氛条件下的热分解行为相比较,poss的残炭率有非常显著的提高(由2.6%增至53.7%),这充分说明在空气气氛中氧气的作用下poss分子结构中的乙烯基团被氧化并参与了凝聚相残炭的形成,大幅提高了poss在空气气氛热重分析中的残炭率。同时,阻燃pp复合材料也表现为两步降解过程,即200~400℃和400~700℃。在第一步降解过程中,pp1样品的t
onset
值和t
max
值均高于纯pp和poss,表明poss的加入可以有效提高pp材料的热稳定性。此外,pp1的残炭率由氮气气氛下的2.9%增加至空气气氛的8.3%,说明在空气气氛条件下poss粒子参与促进凝聚相成炭反应的能力得到了明显增强。与氮气气氛不同,相较于纯pp,pp3和pp4的初始分解温度明显增加,热分解速率亦明显降低,表明在空气气氛条件下pp的大分子链被氧化并参与了成炭过程。第二步降解过程中的重量损失是由在空气气氛下第一降解过程中形成的热不稳定残炭的再氧化降解造成。此外,还发现在所有样品中含有1wt%poss的pp3阻燃材料具有最低的热分解速率,且在700℃时具有9.9%的最高残炭率。这可归因于第一步分解中产生的膨胀炭层形成的物理阻隔层阻止了内部基体树脂的进一步降解。值得注意的是,与n2气氛一样,加入过多的poss粒子(2wt%或更高)也会由于团聚而同样降低样品的最终残炭率。
[0178]
将上述应用例4~5、应用对比例2~4与纯pp(pp0)进行垂直燃烧ul94测试和极限氧指数测试,结果如下表:
[0179][0180]
将上述应用例4、应用对比例3与纯pp(pp0)进行锥形量热测试,结果如下表:
[0181][0182]
上表可以通过使用火灾性能指数(fpi)和火灾增长速率(fgr)进一步分析各种pp复合材料的阻燃性能,其中fpi是点火时间(tti)与phrr的比值,fgr是phrr与t
‑
phrr
的比值。通常,较高的fpi值和较低的fgr值表示更好的阻燃性。如上表所示,纯pp(pp0)和应用对比例3样品(pp2)的火灾性能指数分别为0.024sm2/kw和0.057sm2/kw,火灾增长速率分别为5.69kw/m2s和1.66k w/m2s。与纯pp和pp2相比,应用例4样品(pp3)具有更高的fpi值(0.064sm2/kw)和更低的fgr值(1.26kw/m2s),表明所制备的pp/mppr/poss复合材料具有更好的阻燃性能。烟释放速率(spr)和总烟释放量(tsp),与纯pp相比,pp2和pp3的tsp峰值从20.27m2分别降至19.17m2和16.39m2。此外,pp3的最大烟释放速率(pspr)值降低至0.049m2/s(仅为纯pp的29%)。上述结果表明,mppr/poss复合阻燃剂除对pp的阻燃性能有明显改善作用,还具有优异的抑烟功能:在燃烧过程中,poss分解产生的无机刚性二氧化硅粒子覆盖在炭层的表面,从而有效抑制了材料的燃烧。显然,燃烧过程中pp降解形成的可燃性气体以及碳微粒很难从增强的致密膨胀炭层中逸出,从而可有效减少烟雾的产生。
[0183]
将上述应用例4~5、应用对比例2~4与纯pp(pp0)进行差示扫描量热分析,结果如下表:
[0184][0185]
将上述应用例4~5、应用对比例2~4与纯pp(pp0)进行力学性能测试,结果如下表:
[0186][0187]
将上述应用例4、应用对比例3制备所得样品在锥形量热试验后阻燃pp材料炭层采用eds进行了对比分析,结果如下表:
[0188][0189]
可明显看出,应用例4与应用对比例3之间不同样品炭层中的各元素含量有很大变化,即未引入poss(pp2)时,炭层的c元素含量为47.4%,而加入poss(pp3)后,c元素含量提高到了65.6%。上述结果再次有力说明poss纳米粒子的加入明显促进了pp大分子链参与形成凝聚相炭层的成炭反应,从而提高了pp阻燃材料的残炭率和凝聚相炭层质量。因此,通过利用mppr和适量poss之间强的协效阻燃作用可显著改善凝聚相炭层的质量,提高体系的阻燃性能。
[0190]
应用例6~8、应用对比例5~8
[0191]
本应用例6~8、应用对比例5~8是利用合成例1制备所得单分子膨胀型阻燃剂mppr与聚丙烯、八乙烯基低聚倍半硅氧烷、玻璃纤维及马来酸酐接枝聚丙烯混合制备mppr/
poss/gf复合协效无卤阻燃聚丙烯复合材料,即一种mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料的制备方法,主要包括以下步骤:
[0192]
①
将主要包括单分子膨胀型阻燃剂mppr、聚丙烯、八乙烯基低聚倍半硅氧烷、玻璃纤维及马来酸酐接枝聚丙烯的组分原料混合均匀后作为混合料,通过双螺杆挤出机熔融挤出造粒,收集所得复合材料粒料;其中,双螺杆挤出机的温度设置为160~200℃,挤出速度为100~150r/min;
[0193]
②
将步骤
①
所得复合材料粒料通过平板硫化机热压成型处理,即得mppr/poss/gf复合协效无卤阻燃聚丙烯复合材料;其中,平板硫化机的热压温度为185℃~195℃,压力为10~15mpa。
[0194]
其中,上述制备方法中单分子膨胀型阻燃剂mppr、聚丙烯(pp)、八乙烯基低聚倍半硅氧烷(ov
‑
poss)、玻璃纤维(gf)及马来酸酐接枝聚丙烯(mapp),以重量份数计,应用例6~8、应用对比例5~8为下述配比:
[0195] ppmpprov
‑
possgfmapp应用对比例5(gfpp1)48211300应用对比例6(gfpp2)45211303应用例6(gfpp3)43211305应用例7(gfpp4)40211308应用对比例7(gfpp5)63211105应用例8(gfpp6)53211205应用对比例8(gfpp7)33211405
[0196]
将上述应用例6、8,应用对比例7、8所得样品在氮气气氛下进行热重分析,结果如下表:
[0197][0198]
可以看出,随玻纤含量的增加,阻燃玻纤增强pp样品的残炭率和起始分解温度(t
onset
)均逐渐增加,说明玻纤的加入提高了阻燃pp材料的热稳定性。其中,残炭率的增加主要是因为玻纤为不燃物,在升温过程中几乎不发生降解,残炭的主要成分为炭化玻纤,故残炭率会随着gf含量的增加而增加。gf含量为10wt%时,pp/mppr/poss/gf复合材料的t
onset
值为281.5℃,最大热降解速率所对应的温度(t
max
)为457.9℃;与其相比,玻纤添加量为30wt%时,复合材料的初始分解温度提高到287.7℃,最大热降解速率明显降低,且具有最高的t
max
值(462.1℃)。这可能是因为玻纤的加入及适当提高其负载量有利于提高阻燃pp材料在燃烧过程中形成的炭层强度,起到了有效隔绝热氧传递的作用,从而在一定程度上抑
制了材料的降解,有利于阻燃性能的改善。
[0199]
将上述应用例6、8,应用对比例7、8所得样品进行垂直燃烧ul94测试和极限氧指数测试,结果如下表:
[0200][0201]
上表给出了玻纤含量从10wt%增至40wt%时,阻燃玻纤增强pp复合材料的极限氧指数(loi)和垂直燃烧ul94等级的变化情况。从表中可以看出,随着gf含量的提高,随玻纤含量的提高,材料阻燃性能呈提高趋势:gfpp5的loi值为27.5%,可通过ul
‑
94v
‑
1(3.2mm)等级,gfpp6和gfpp3均能通过ul
‑
94v
‑
0等级,但gfpp3具有更高的loi值。这主要有两方面原因:一方面,提高玻纤含量降低了基体树脂聚丙烯的含量,相当于减少了阻燃材料中的可燃物含量,且阻燃体系中含硅poss原位分解产生的纳米sio2粒子通过吸附于玻纤表面,可达到抑制玻纤的“烛芯效应”目的;另一方面,玻纤的加入有助于提高阻燃体系中膨胀型阻燃剂mppr分解产生的膨胀炭层结构的强度,从而更好地阻隔外界氧气和热量与基体树脂燃烧降解区之间的接触,提高材料的阻燃性能。但当玻纤含量增至40wt%以上时,由于玻纤在基体树脂中难以分散,使得mppr和poss分解分别产生的膨胀炭层和sio2颗粒不能全部覆盖于玻纤表面,因而导致玻纤的“烛芯效应”无法得到完全抑制,此时材料亦难以自熄无法通过垂直燃烧ul94 v
‑
0级别。
[0202]
将上述应用例6、8,应用对比例7、8所得样品通过微型燃烧量热仪进行测试,结果如下表:
[0203][0204]
将上述应用例6、8,应用对比例7、8所得样品的力学性能进行测试,结果如下表:
[0205][0206]
从上表可以看出,随着玻纤含量的增加,gfpp复合材料的断裂伸长率稍呈下降趋势,这是因为刚性玻纤的加入稀释了基体树脂的浓度,从而会一定程度降低pp树脂的断裂伸长率。从表中还可以看出,阻燃pp复合材料的拉伸、弯曲和缺口冲击强度均随玻纤含量的增加而增加,且玻纤含量为30%时,相应复合材料的力学性能达到最佳,进一步增加玻纤含量,阻燃pp复合材料的力学性能反而下降。这是因为玻纤含量较低时,pp基体占比较多,外界应力主要作用在基体树脂上,所以阻燃材料强度相对较低。而玻纤添加量达到40wt%时,过多的玻纤在基体树脂中难以分散,且不能很好地被阻燃pp基体树脂包覆,从而导致其力学性能下降。因此,玻纤添加量为30wt%时,阻燃pp材料的力学性能最佳。
[0207]
将上述应用例6、7,应用对比例5、6所得样品的进行阻燃性能和力学性能测试,结果如下表:
[0208][0209]
可以看出,马来酸酐接枝聚丙烯(mapp)的加入可以显著提高mppr/poss阻燃玻纤增强pp复合材料的力学性能。随mapp含量的增加,材料的拉伸强度、缺口冲击强度和弯曲强度呈明显提高趋势,但当mapp含量进一步从5wt%(gfpp3)增至8wt%时(gfpp4),阻燃玻纤增强pp材料的力学性能变化不大(拉伸和弯曲强度小幅上升,缺口冲击强度略有下降),说明加入的mapp只有在适当含量范围内才能有效提高材料的力学性能。这是因为加入过多的mapp相当于一定程度降低了玻纤的含量,因而会减弱玻纤对体系的增强作用。此外,随着mapp含量从0.0wt%增至8.0wt%,阻燃玻纤增强pp材料的熔融指数从4.1g/10min提高至9.3g/10min,材料的熔体流动性明显改善,这也有利于改善体系的加工和成型性能。从上表
还可以看出,mapp的加入及其含量的适当增加,阻燃玻纤增强pp材料的阻燃性能稍有改善,可归因于体系流动性提高在一定程度上改善了阻燃剂的分散性。而过多加入mapp后(尤其是超过8wt%)体系的界面结合和相容性明显下降,且玻纤和阻燃剂填料还发生一定程度团聚,故相容剂mapp的最优添加量为5wt%左右。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1