一种含二氧戊环的三唑类化合物及其中间体的制备方法与流程
1.本发明属于有机合成技术领域,具体涉及一种含二氧戊环的三唑类化合物及其中间体的制备方法。
背景技术:
2.三唑类化合物具有广谱的杀菌活性,由于其具有内吸功能和保护、治疗作用而被广泛用于子囊菌、担子菌等真菌引起的多种病害的防治。麦角甾醇是许多菌类细胞膜的主要甾醇,三唑类化合物通过氮杂环上的氮原子与细胞色素中的铁离子结合,从而阻断了构成菌类细胞膜的麦角甾醇的合成,从而抑制了细胞膜的合成,达到杀菌的效果。
3.三唑类化合物如丙环唑、苯醚甲环唑等均具有良好的杀菌活性。目前合成含二氧戊环的三唑类化合物的路线主要有:1)环合
‑
溴代
‑
缩合法:;2)溴代
‑
环合
‑
缩合法:;3)溴代
‑
缩合
‑
环合法:;
4)氯代苯乙酮经水解、酯化、取代、环合合成法:。
4.但上述路线均存在副产物多的问题,由于其缩合反应是一个亲核取代反应,1,2,4
‑
三唑与相应的卤代芳基乙酮在缚酸剂的存在下进行缩合反应的收率总是不够令人满意,且存在反应时间长(回流条件下反应至少10小时)而导致生产成本高、环境不友好的问题。另外还存在以下问题:1)溴化反应中使用溴素进行溴代,原子利用率仅为1/2,反应同时产生了溴化氢气体或溴化废水,酸性强,难以处理;2)缩合反应的1,2,4
‑
三氮唑反应后产生15%左右的产品异构体,导致分离纯化难、收率低;3)产品精制通常采用硝酸成盐法,产生大量废水;采用蒸馏结晶法,蒸馏需要高温300℃左右,存在较大的安全隐患,高温使产品分解成杂质,造成收率降低。
5.因此,迫切希望有一种新的含二氧戊环的三唑类化合物及其中间体的制备方法,能解决当前合并收率低、反应时间长、生产成本高、操作复杂的问题。
技术实现要素:
6.本发明针对含二氧戊环的三唑类化合物制备过程中由于溴化反应中使用溴素进行溴代而造成的原子利用率低及酸性废气废水产生的问题,以及缩合反应收率低、副产物难分离、产品精制过程中能耗高、易产生杂质的问题,提出了全新的合成路线,其主要优点是能够避免使用溴素及减少中间产物异构体产生,具有环境友好、对设备要求低、反应路线短及操作安全的优点。
7.本发明为了实现上述技术目的,提供如下技术方案:一种三唑类化合物的制备方法,使式(v)所示化合物与式(iv)所示化合物在路易斯酸存在下反应,制备得到如式(iii)所示化合物:式(iii)及式(v)中,r1选自卤素、芳氧基或卤代芳氧基。
8.进一步地,所述路易斯酸为无水氯化铁、无水氯化铝、三氟化硼、四氯化钛、五氟化锑中的任一种或任几种的混合物。
9.进一步地,溶剂为二氯甲烷、氯仿、四氯化碳、1,2
‑
二氯乙烷、环己烷、甲基环己烷、环戊烷中的任一种或任几种的混合物。
10.进一步地,r1选自f、cl、br、苯氧基、对氯苯氧基、对溴苯氧基、对氟苯氧基、间氯苯氧基、间溴苯氧基、间氟苯氧基、邻氯苯氧基、邻溴苯氧基或邻氟苯氧基;再进一步地,r1为cl或对氯苯氧基。
11.进一步地,反应温度为0
‑
30℃;再进一步地,反应温度为0
‑
20℃;再进一步地,反应温度为5
‑
10℃。
12.本发明还公开了一种含二氧戊环的三唑类化合物的制备方法,根据如上所述的制备三唑类化合物的方法制备得到式(iii)化合物,与式(ii)化合物反应,制备得到如式(i)所示化合物:式(iii)及式(i)中,r1选自卤素、芳氧基或卤代芳氧基;r2选自h或c1‑6烷基。
13.进一步地,溶剂为甲苯或者环己烷。
14.进一步地,式(iii)化合物与式(ii)化合物在对甲苯磺酸存在下升温至回流,分水,至水完全分尽后进行后处理。
15.进一步地,所述后处理为冷却至室温,水洗分层,有机相脱溶后加入结晶溶剂进行结晶;进一步地,所述结晶溶剂为叔丁基甲醚、异丙醚、乙醇、甲苯、环己烷、甲基环己烷中的任一种或任几种的混合物;进一步地,r1选自f、cl、br、苯氧基、对氯苯氧基、对溴苯氧基、对氟苯氧基、间氯苯氧基、间溴苯氧基、间氟苯氧基、邻氯苯氧基、邻溴苯氧基或邻氟苯氧基,r2选自h、me、et、n
‑
pr、i
‑
pr、n
‑
bu、i
‑
bu、t
‑
bu、s
‑
bu;进一步地,r1为cl或对氯苯氧基,r2为h、me、et或n
‑
pr;再进一步地,r1为对氯苯氧基,r2为me;或r1为cl,r2为h;或r1为cl,r2为et;或r1为cl,r2为n
‑
pr。
16.在本申请中所使用的专业术语或英文缩写均具有本领域技术人员公知的含义,具体如下:h为氢,me为甲基,et为乙基,pr为丙基,bu为丁基,n表示“正”,i表示“异”,s表示“仲”,t表示“叔”,hplc为高效液相色谱,dmf为n,n
‑
二甲基甲酰胺。
17.由于采用了以上技术,本发明与现有技术相比,其显著优点为:1)工艺路线简便,反应步骤少,工艺简单,生产成本低;2)避免使用溴素,具有环境友好、绿色安全的优点;3)通过1h
‑
1,2,4
‑
三唑
‑1‑
乙酸引入三氮唑基团,避免了异构体1,3,4
‑
三氮唑副产物的产生,提高了反应收率;4)产物后处理仅需通过简单的溶剂结晶工艺进行,无需采用硝酸成盐法和高温蒸馏法,在提高产品收率和含量的情况下降低对设备的要求并减少成本,适合工业化生产。
附图说明
18.图1为实施例2步骤2)的中控高效液相色谱图;图2为对比例步骤2)的中控高效液相色谱图。
具体实施方式
19.为了使本发明的技术方案及优点更加清楚明白,以下结合具体的实施例对本发明进行进一步详细说明,但并不因此而限定本发明的范围。
20.实施例11)(2,4
‑
二氯苯基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮的合成:将2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯85g溶于1,2
‑
二氯乙烷200g。向四口瓶内加入无水氯化铝82g,1,2
‑
二氯乙烷300g,再加入间二氯苯75g,控制温度为0
‑
5℃。
21.将配置好的2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯的1,2
‑
二氯乙烷溶液滴加至四口瓶中,滴加温度控制在5
‑
10℃,约4h滴加完毕,滴完保温2h,反应合格后,将四口瓶内的物料向300g水中滴加,滴加完毕后,分液得到油相,将油相再用300g水水洗,分液得到油相,负压脱溶出1,2
‑
二氯乙烷,得到(2,4
‑
二氯苯基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮粗品,再用乙醇重结晶,干燥,得到(2,4
‑
二氯苯基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮产品129.7g,含量98.2%,收率为97.5%。
22.2)1
‑
[2
‑
(2,4
‑
二氯苯基)
‑4‑
丙基
‑
1,3
‑
二氧戊环
‑2‑
甲基]
‑
1氢
‑
1,2,4三唑的合成:向四口瓶内加入上步得到的(2,4
‑
二氯苯基)
‑2‑
(1h
‑
1,2,4
‑
三氮唑
‑1‑
基)
‑1‑
乙酮125g,再加入环己烷500g,催化剂对甲苯磺酸,1,2
‑
戊二醇65g,升温至回流,回流反应8h,反应合格后,向反应中加入300g水进行水洗,分层,油相负压脱溶,得到1
‑
[2
‑
(2,4
‑
二氯苯基)
‑4‑
丙基
‑
1,3
‑
二氧戊环
‑2‑
甲基]
‑
1氢
‑
1,2,4三唑即丙环唑163.6g,含量99.2%,收率为98.9%。
[0023]
实施例21)1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮的合成:
将2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯61.5g溶于二氯甲烷200g。向四口瓶内加入无水氯化铝62g,二氯甲烷400g,再加入3,4
’‑
二氯
‑
二苯醚98.5g,控制温度为10
‑
15℃。
[0024]
将配置好的2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯的二氯甲烷溶液滴加至四口瓶中,滴加温度控制在15
‑
20℃,约4h滴加完毕,滴完保温2h,反应合格后,将四口瓶内的物料向500g水中滴加,滴加完毕后,分液得到油相,将油相再用500g水水洗,分液得到油相,脱溶出二氯甲烷,得到1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮粗品,再用石油醚重结晶,干燥,得到1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮产品141.2g,含量98.6%,收率为97.1%。
[0025]
2)顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚的合成:向四口瓶内加入上步得到的1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮72g,再加入环己烷400g,对甲苯磺酸,1,2
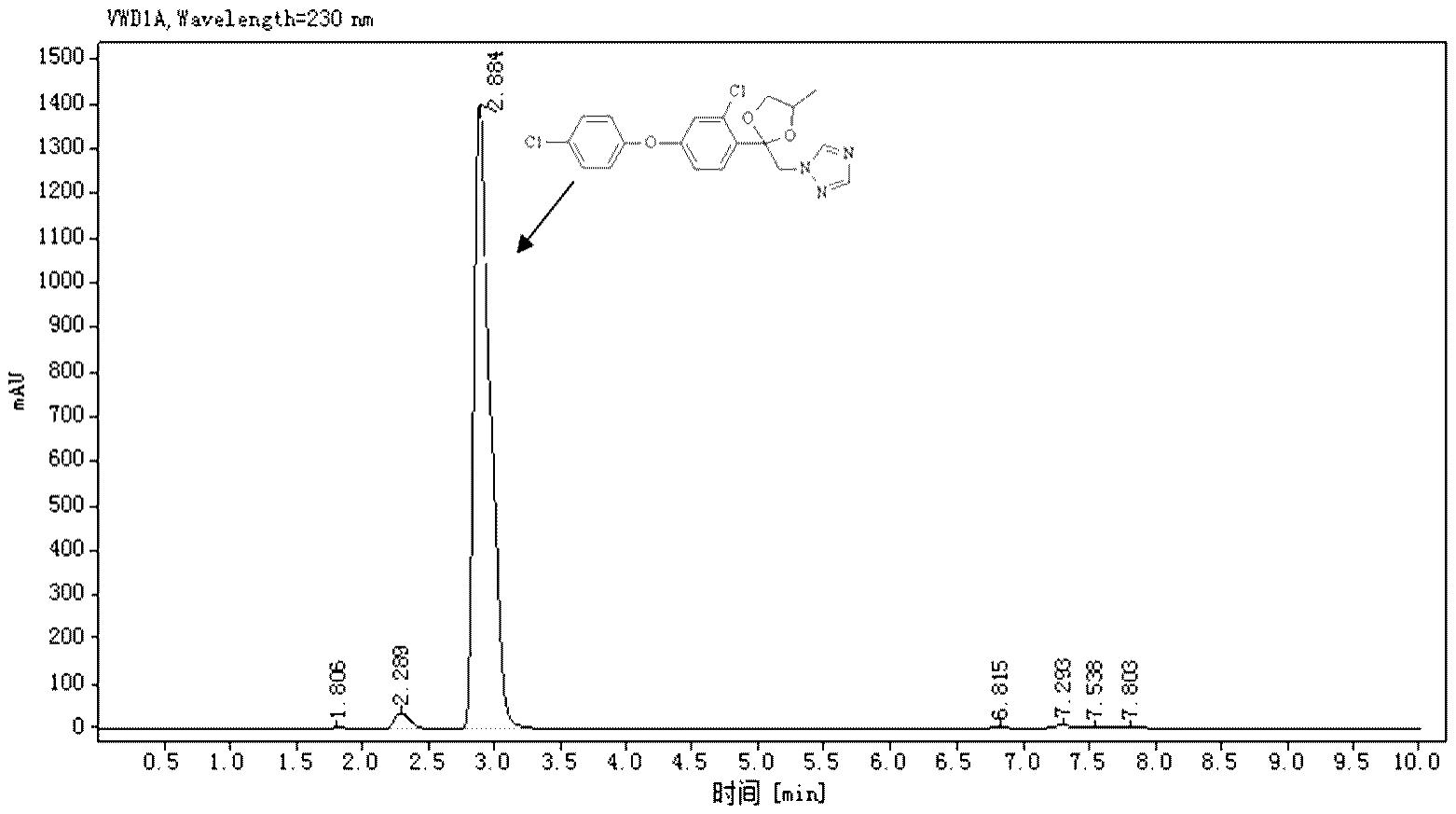
‑
丙二醇20g,升温至回流,回流反应6h,进行中控检测,hplc图见附图1。
[0026]
反应合格后,向反应中加入200g水进行水洗,分层,油相负压脱溶,得到顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚粗品,再用异丙醚进行重结晶,干燥,得到顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚产品(即苯醚甲环唑)82.2g,含量98.6%,收率为97.8%。
[0027]
实施例31)1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮的合成:将2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯61.5g溶于1,2
‑
二氯乙烷200g。向四口瓶内加入无水氯化铝76g,1,2
‑
二氯乙烷400g,再加入3,4
’‑
二氯
‑
二苯醚98.5g,控制温度为10
‑
15℃。
[0028]
将配置好的2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯的1,2
‑
二氯乙烷溶液滴加至四口瓶中,滴加温度控制在15
‑
20℃,约5h滴加完毕,滴完保温2h,反应合格后,将四口瓶内的物料向400g水中滴加,滴加完毕后,分液得到油相,将油相再用400g水水洗,分液得到油相,脱溶出1,2
‑
二氯乙烷,得到1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮粗品,再用石油醚重结晶,干燥,得到1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙
酮产品141.5g,含量98.5%,收率为97.2%。
[0029]
2)顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚的合成:向四口瓶内加入上步得到的1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮72g,再加入甲苯400g,对甲苯磺酸,1,2
‑
丙二醇20g,升温至回流,回流反应6h。
[0030]
反应合格后,向反应中加入200g水进行水洗,分层,油相负压脱溶,得到顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚粗品,再用异丙醚/乙醇进行重结晶,干燥,得到顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚产品(即苯醚甲环唑)82.6g,含量98.4%,收率为98.2%。
[0031]
实施例41)1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮的合成:将2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯61.5g溶于二氯甲烷200g。向四口瓶内加入无水氯化铝76g,二氯甲烷400g,再加入3,4
’‑
二氯
‑
二苯醚98.5g,控制温度为10
‑
15℃。
[0032]
将配置好的2
‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑
乙酰氯的二氯甲烷溶液滴加至四口瓶中,滴加温度控制在15
‑
20℃,约5h滴加完毕,滴完保温2h,反应合格后,将四口瓶内的物料向300g水中滴加,滴加完毕后,分液得到油相,将油相再用300g水水洗,分液得到油相,脱溶出二氯甲烷,得到1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮粗品,再用甲基环己烷重结晶,干燥,得到1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮产品141.8g,含量98.6%,收率为97.5%。
[0033]
2)顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚的合成:向四口瓶内加入上步得到的1
‑
(2
‑
氯
‑4‑
(4
‑
氯苯氧基)
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑1‑
乙酮72g,再加入环己烷400g,对甲苯磺酸,1,2
‑
丙二醇21g,升温至回流,回流反应6h。
[0034]
反应合格后,向反应中加入200g水进行水洗,分层,油相负压脱溶,得到顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚粗品,再用异丙醚/甲基环己烷进行重结晶,干燥,得到顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚产品(即苯醚甲环唑)82.7g,含量97.9%,收率为97.7%。
[0035]
对比例1)3,4
’‑
二氯
‑
二苯醚的合成:向四口瓶内加入间二氯苯,对氯苯酚和dmf,投入碳酸钾和氯化亚铜,进行回流反
应,反应合格后,抽滤,滤液负压脱溶回收溶剂,得到粗品,再蒸馏收集馏分得到产品,含量97.1%,收率91.2%。
[0036]
2)2
‑
氯
‑4‑
(4
‑
氯苯氧基)苯乙酮的合成:向四口瓶内加入3,4
’‑
二氯
‑
二苯醚,1,2
‑
二氯乙烷,无水三氯化铝,滴加乙酰氯,加完后保温4h,反应合格后与水进行水解水洗,油相负压脱溶,加入石油醚进行结晶,得到产品,含量97.4%,收率90.1%。
[0037]
3)2
‑
甲基
‑2‑
[2
‑
氯
‑
4(4
‑
氯苯氧基)苯基]
‑4‑
甲基
‑
1,3
‑
二噁戊烷的合成:向四口瓶内加入环己烷,加入到反应釜,加入2
‑
氯
‑4‑
(4
‑
氯苯氧基)苯乙酮和1,2
‑
丙二醇,加入对甲苯磺酸,升温回流分水,合格后直接用于下一步反应。
[0038]
4)2
‑
溴甲基
‑2‑
[2
‑
氯
‑
4(4
‑
氯苯氧基 )苯基]
‑4‑
甲基
‑
1,3
‑
二噁戊烷的合成:向四口瓶内加入上一步物料,滴加液溴,加完后搅拌1h,反应合格后,加入水洗涤,油相负压脱溶回收环己烷,得到产品,含量95.2%,收率为97.8%。
[0039]
5)顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚的合成:向四口瓶内加入2
‑
溴甲基
‑2‑
[2
‑
氯
‑
4(4
‑
氯苯氧基)苯基]
‑4‑
甲基
‑
1,3
‑
二噁戊烷,dmf,再加入1,2,4
‑
三氮唑和碳酸钾,升温反应10h,进行中控检测,hplc图见附图2。合格后升温进行负压蒸馏dmf,蒸馏完溶剂后,降温加入二甲苯和水,搅拌0.5小时溶解后分液得到油相,负压脱溶回收甲苯,得顺,反
‑3‑
氯
‑4‑
[4
‑
甲基
‑2‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基甲基)
‑
1,3
‑
二噁戊烷
‑2‑
甲基]苯基
‑4‑
氯苯基醚即苯醚甲环唑粗品。苯醚环唑粗品通过成盐法与硝酸成硝酸盐,过滤后再进行碱解,得到苯醚甲环唑苯油。将苯油使用甲基叔丁基醚与异丙醚结晶,得到苯醚甲环唑,含量93.2%,收率75.6%。
[0040]
上述实施例仅为本发明较佳的具体实施方案,不应视为对于本发明的限制,本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围,任何熟悉本技术领域的技术人员在本发明披露的技术范围之内,根据本发明的技术方案及其发明构思进行等同替换或改进,都涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1