一种特殊支链伯醇烷基糖苷的制备方法与流程
1.本发明属于精细化工表面活性剂技术领域,具体地说,涉及一种特殊支链伯醇烷基糖苷的制备方法。
背景技术:
2.特殊支链伯醇,又称β
‑
支链醇,具有低粘度、低凝点、良好的生物降解能力、低色泽和优异的热稳定性等特殊性能而被广泛用于润滑剂、表面活性剂等领域。
3.以特殊支链伯醇合成的特殊支链烷基糖苷,不仅保留了烷基糖苷(apg)的独特性能,而且在高浓度下也可保持液体状态和流动性。低烷基链的支链apg兼有耐高浓度强碱、高浓度电解质和高温等特殊功能,在轻纺、冶金和采油等领域可作为耐极端环境的特种功能助剂使用,起到增溶、润湿、渗透、乳化和清洁等作用,目前研究还是较少[杨秀全,张军,周媛,等.烷基糖苷及其衍生物的技术进展[j].日用化学工业,2012,42(3):213
‑
219;白亮,杨秀全,吴志宇等.支链烷基糖苷的合成及性能研究[j].日用化学工业,2015,45(10):557
‑
560]。
[0004]
支链醇羟基的空间位阻大,导致其与葡萄糖生成糖苷的反应活性低,使用传统的单一酸性催化剂,反应转化率很低甚至不转化。通过加强反应条件如升高温度,延长反应时间或提高体系真空度,效果均不理想。
[0005]
公开号为cn109939612a的专利申请公开了一种格尔伯特醇烷基糖苷表面活性剂及其制备方法,其包括步骤:格尔伯特醇与葡萄糖在酸性复合相转移催化剂作用下,加热并抽真空回流进行缩醛反应生成格尔伯特醇烷基糖苷,用碱中和后即得;酸性复合相转移催化剂包括a、b两个组分,a组分选自磺酸类化合物和硫酸,b组分选自阳离子表面活性剂、环糊精,聚乙二醇和甲基封端聚乙二醇。本发明开发了一种酸性复合相转移催化体系,可提高特别是长碳链格尔伯特醇和葡萄糖缩合成烷基糖苷的反应活性,通过所述方法葡萄糖和格尔伯特醇形成缩醛即生成格尔伯特醇烷基糖苷。本发明的方法工艺流程简洁,副反应少,产品活性含量高,但其反应温度高、反应时间较长和选择性不太理想。
[0006]
公开号为cn102643312a的专利申请的技术方案主要是用直接苷化法来合成异构伯醇烷基糖苷,以异构伯醇,葡萄糖为原料,以路易斯(lewis)酸为催化剂,葡萄糖在酸性催化剂存在条件下可失去一分子水,形成碳正离子,然后再与一分子的异构伯醇作用形成稳定的缩醛即生成粗产品的异构伯醇烷基糖苷。该方法对位阻较大的2
‑
丙基庚醇和2
‑
丁基辛醇的效果不佳。
[0007]
公开号为cn1105667a的专利申请公开了一种烷基葡糖苷的相转移催化合成法,反应与分馏并行操作,直接产物为淡黄色,不需脱色即可满足使用要求。它采用了包括所有在醚化反应中所用的酸性催化剂;将聚乙二醇用作相转移催化剂;使用c3
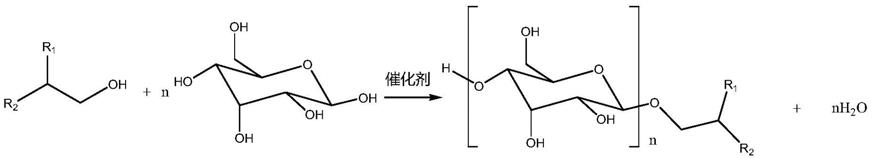
‑
c5的一元醇作为活化溶剂;使用乙醇作为烷基葡糖苷产物气味的改善剂;使用na2so3与无机碱或有机碱组成的二元中和剂;所用的糖类物质可以是各种单糖、双糖及多糖。该方法使用的是短链一元醇作为“活化试剂”,相当于“两步法”即短链糖苷转化法,方法有异味以及短碳链糖苷残留问题
致产品质量和表面活性降低,应用上受限于低端领域。
技术实现要素:
[0008]
针对现有技术中特殊支链伯醇与葡萄糖缩醛生成糖苷的反应活性不高、转化率低、反应时间长、副反应多的问题,本发明的目的是提供一种特殊支链伯醇烷基糖苷的制备方法,该方法采用了有机酸/二元复合相转移催化剂体系,有效提高了特殊支链伯醇葡萄糖缩醛的反应活性,该方法工艺流程简洁,反应时间短,糖苷转化率高且副反应少,而且该方法制备的特殊支链伯醇烷基糖苷具有表面张力低、润湿性能优异、低泡的特点。
[0009]
为了实现上述目的,本发明采用的技术方案如下:
[0010]
本发明的第一方面提供了一种特殊支链伯醇烷基糖苷的制备方法,包括以下步骤:
[0011]
将摩尔比为(2~12):1的特殊支链伯醇与葡萄糖混合,加入有机酸/二元复合相转移催化剂,有机酸/二元复合相转移催化剂与特殊支链伯醇和葡萄糖的总质量比为1:(40~300)(优选为1:(150~285)),加热混合物并且抽真空至残压小于50mmhg为止,在温度为90~120℃的条件下回流4~24小时,当混合物变为半透明到透明状后监测反应至残余葡萄糖小于等于0.5%时,用氢氧化钠溶液中和,经真空减压脱醇,然后加入去离子水制成固含量为60%以上的溶液,经过脱色步骤,获得所述特殊支链伯醇烷基糖苷溶液;
[0012]
所述有机酸/二元复合相转移催化剂是由质量比为(0.2~5):1(优选为(0.2~2):1)的a组分和b组分制成,所述a组分为磺酸类化合物,所述磺酸类化合物选自对甲基苯磺酸(ptsa)、苯磺酸(bsa)、乙基苯磺酸(ebsa)中的一种,所述b组分是由质量比为(0.1~5):1的b1组分和b2组分制成,所述b1组分选自四丁基溴化铵(tbab)、四丁基氯化铵(tbac)中的一种;所述b2组分选自三甲基苄基氯化铵(tmbac)、三辛基甲基氯化铵(tomac)、三乙基苄基溴化铵(tebab)、三乙基苄基氯化铵(tebac)中的一种;
[0013]
所述真空减压脱醇的步骤如下:将混合液转移至蒸馏装置脱醇,在体系压力小于40mmhg时,温度为100
‑
180℃的条件下蒸馏,至气相温度降至35℃以下时脱醇结束;
[0014]
所述脱色步骤如下:加入漂白剂和漂白助剂进行脱色,所述漂白剂为双氧水(h2o2)、次氯酸钠(naclo)、亚硫酸钠(na2so3)中的至少一种,所述漂白助剂为氧化镁(mgo);所述漂白助剂的加入量为烷基糖苷有效质量的0.05
‰
~0.3
‰
(优选为0.15
‰
~0.3
‰
),所述漂白剂的加入量为烷基糖苷有效质量的0.5~4%(优选为2%)。
[0015]
所述特殊支链伯醇与葡萄糖的摩尔比为(2~5):1。
[0016]
所述特殊支链伯醇选自2
‑
丙基庚醇、2
‑
丁基辛醇等中的至少一种。
[0017]
所述葡萄糖为无水葡萄糖。
[0018]
所述特殊支链伯醇烷基糖苷溶液中,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.30~1.65。
[0019]
所述特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为(50~85):1。
[0020]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0021]
本发明提供的特殊支链伯醇烷基糖苷的制备方法,以特殊支链伯醇、葡萄糖为原料,在有机酸/二元复合相转移催化剂体系下,缩醛反应生成特殊支链伯醇烷基糖苷;采用
有机酸/二元复合相转移催化剂体系,有效提高了特殊支链伯醇与葡萄糖缩醛的反应活性,该方法工艺流程简洁,糖苷转化率高且副反应少,所得的溶液中烷基糖苷含量高,具有表面张力低、润湿性性能优异、低泡等特点,可用于金属清洗、工业清洗、纺织助剂等领域,发展应用前景广阔。
具体实施方式
[0022]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0023]
本发明实施例的特殊支链伯醇烷基糖苷的反应式如下:
[0024][0025]
式中,r1代表碳数为2~6的直链烷基,r2独立地代表碳数为3~9的直链或异构的烷基;n≥1的整数。
[0026]
实施例1
[0027]2‑
丙基庚醇烷基糖苷的合成
[0028]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.0mol,316.4g),无水葡萄糖(0.5mol,90g),在搅拌下加入ebsa(0.0012mol,0.80g),tbab(0.0012mol,0.40g)和tebac(0.00175mol,0.40g),加热混合物并且抽真空至残压35mmhg,在108℃下回流10小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.09%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在107~176℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.42%,加去离子水溶解,配成含有固含量为70%的水溶液,加入相对于烷基糖苷有效质量的0.2
‰
的mgo和2%的双氧水进行脱色,即得到橙色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量1.0%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.45。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为70:1。
[0029]
实施例2
[0030]2‑
丙基庚醇烷基糖苷的合成
[0031]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(1.5mol,237.3g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00377mol,0.65g),tbac(0.0023mol,0.65g)和tebac(0.00058mol,0.16g)的复合催化剂,加热混合物并且抽真空至残压35mmhg,在108℃下回流7.5小时,当混合物变为半透明到透明状后监测反应至残余葡萄糖为0.03%,加入2.2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在106~176℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.27%,加去离子水溶解,配成固含量70%的水溶液,加入
相对于烷基糖苷有效质量0.2
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量1.2%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.47。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为60:1。
[0032]
实施例3
[0033]2‑
丙基庚醇烷基糖苷的合成
[0034]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.0mol,316.4g),无水葡萄糖(0.5mol,90g),在搅拌下加入bsa(0.005mol,0.81g),tbab(0.0025mol,0.81g)和tebab(0.00147mol,0.40g)的复合催化剂,加热混合物并且抽真空至残压25mmhg,在105℃下回流10小时,当混合物变为半透明到透明状后监测反应至残余葡萄糖为0.06%,加入2.0g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度至108~175℃左右,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.30%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.15
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量0.83%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.41。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为82:1。
[0035]
实施例4
[0036]2‑
丙基庚醇烷基糖苷的合成
[0037]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.0mol,316.4g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00377mol,0.65g),tbab(0.00124mol,0.40g)和tebab(0.0012mol,0.40g)的复合催化剂,加热混合物并且抽真空至残压35mmhg,在108℃下回流10小时,当混合物变为半透明到透明状后监测反应至残余葡萄糖为0.02%,加入2.0g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度至112~177℃左右,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.30%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.15
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量0.94%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.47。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为73:1。
[0038]
实施例5
[0039]2‑
丙基庚醇烷基糖苷的合成
[0040]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(1.5mol,237.3g),无水葡萄糖(0.5mol,90g),在搅拌下加入ebsa(0.00435mol,0.81g),tebac(0.00356mol,0.81g)和tbac(0.0036mol,0.10g),加热混合物并且抽真空至残压25mmhg,在105℃下回流10小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.02%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在110~175℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.56%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖
苷有效质量0.3
‰
的mgo和2%的na2so3进行脱色,即得到橙色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量1.23%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.40。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为61:1。
[0041]
实施例6
[0042]2‑
丙基庚醇烷基糖苷的合成
[0043]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.5mol,395.5g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00842mol,1.45g)、tebac(0.0021mol,0.48g)和tbac(0.000864mol,0.24g),加热混合物并且抽真空至残压35mmhg,在90℃下回流18小时,当混合物由变为半透明后监测反应至残余葡萄糖为0.50%,加入3.0g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在112~175℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.48%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.3
‰
的mgo和2%的na2so3进行脱色,即得到橙色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量1.30%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.32。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为52:1。
[0044]
实施例7
[0045]2‑
丁基辛醇烷基糖苷的合成
[0046]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(2.5mol,465.8g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00639mol,1.1g),tebac(0.00483mol,1.1g)和tbab(0.00171mol,0.55g),加热混合物并且抽真空至残压20mmhg,在105℃下回流12小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.32%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在120~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.72%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.25
‰
的mgo和2%的naclo进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液。测定其中的多糖的含量1.25%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.44。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为55:1。
[0047]
实施例8
[0048]2‑
丁基辛醇烷基糖苷的合成
[0049]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(2.5mol,465.8g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00534mol,0.92g),tmbac(0.00495mol,0.92g)和tbab(0.00071mol,0.23g),加热混合物并且抽真空至残压25mmhg,在108℃下回流10小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.04%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在118~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.77%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.2
‰
的mgo和2%的双氧水进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液。测定其中的多糖的含量0.90%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.47。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为76:1。
[0050]
实施例9
[0051]2‑
丁基辛醇烷基糖苷的合成
[0052]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(1.5mol,279.5g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00430mol,0.74g),tebac(0.00325mol,0.74g)和tbac(0.00266mol,0.74g),加热混合物并且抽真空至残压25mmhg,在108℃下回流9小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.03%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在120~179℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.77%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.2
‰
的mgo和2%的双氧水进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液。测定其中的多糖的含量1.28%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.51。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为54:1。
[0053]
实施例10
[0054]2‑
丁基辛醇烷基糖苷的合成
[0055]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(2.0mol,372.6g),无水葡萄糖(0.5mol,90g),在搅拌下加入bsa(0.00534mol,0.92g),tebac(0.00202mol,0.46g)和tbab(0.00285mol,0.92g),加热混合物并且抽真空至残压25mmhg,在108℃下回流11小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.02%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在121~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.69%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.2
‰
的mgo和2%的naclo进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液。测定其中的多糖的含量0.91%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.47。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为75:1。
[0056]
实施例11
[0057]2‑
丁基辛醇烷基糖苷的合成
[0058]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(1.5mol,279.5g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00215mol,0.37g),tebab(0.00272mol,0.74g)和tbac(0.00133mol,0.37g),加热混合物并且抽真空至残压30mmhg,在115℃下回流7小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.04%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在120~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.67%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.3
‰
的mgo和2%的双氧水进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液。测定其中的多糖的含量1.05%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.61。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为65:1。
[0059]
对比例1
[0060]2‑
丙基庚醇烷基糖苷的合成
[0061]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.0mol,
316.4g),无水葡萄糖(0.5mol,90g),在搅拌下加入ebsa(0.0012mol,0.80g),加热混合物并且抽真空至残压35mmhg,在108℃下回流10小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为11.7%,继续回流6小时,颜色加深,有部分炭化,测定残余葡萄糖为8.1%,继续回流3小时,颜色已变很深,炭化量加大,测定残余葡萄糖为4.3%。停止反应,说明生成糖苷很少,葡萄糖大部分未反应或转化为多糖。加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在107~176℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.48%,加去离子水溶解,配成固含量为60%的水溶液,加入相对于烷基糖苷有效质量的0.2
‰
的mgo和2%的双氧水进行脱色,即得到橙色的2
‑
丙基庚醇烷基糖苷溶液,测定其中的多糖含量9.6%。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为5.3:1。
[0062]
对比例1同实施例1相比,只采用了磺酸类化合物作为催化剂,反应较难转化,且较多的葡萄糖转化为多糖,即反应转化为烷基糖苷的选择性差。
[0063]
对比例2
[0064]2‑
丁基辛醇烷基糖苷的合成
[0065]
在装有搅拌器、温度计、分水装置的四口烧瓶中加入2
‑
丁基辛醇(2.5mol,465.8g)、无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00639mol,1.1g),加热混合物并且抽真空至残压20mmhg为止,在温度为105℃的条件下回流12小时,混合物由白色乳化状变为黄色乳化物监测反应至残余葡萄糖为13.4%,继续回流5小时,乳化物颜色加深,炭化物增多,监测反应至残余葡萄糖为8.8%,提高真空至残压25mmhg继续回流5小时,乳化物颜色变很深,炭化物进一步增多,监测反应至残余葡萄糖为6.3%,停止反应。加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在120~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.72%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.25
‰
的mgo和2%的naclo进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液,测定其中的多糖含量为8.3%。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为7.4:1。
[0066]
对比例2同实施例7相比,只采用了对甲基磺酸类化合物作为催化剂,反应较难转化,且较多的葡萄糖转化为多糖,即反应转化为烷基糖苷的选择性变差。
[0067]
对比例3
[0068]2‑
丙基庚醇烷基糖苷的合成
[0069]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.0mol,316.4g),无水葡萄糖(0.5mol,90g),在搅拌下加入bsa(0.005mol,0.81g)和tbab(0.0025mol,0.81g)的复合催化剂,加热混合物并且抽真空至残压25mmhg,在105℃下回流10小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.23%,继续反应2小时,测定残余葡萄糖为0.08%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在110~175℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.36%,加去离子水溶解,配成固含量为60%的水溶液,加入相对于烷基糖苷溶液0.15
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量7.2%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.52。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷
与多糖的比例为7.3:1。
[0070]
对比例3同实施例3相比,只采用了磺酸类化合物a组分和相转移催化剂中b1组分作为催化剂,所得结果表明转化为烷基糖苷的选择性较差,且反应稍变慢,说明对比例3不能取得本技术的技术效果。
[0071]
对比例4
[0072]2‑
丁基辛醇烷基糖苷的合成
[0073]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(2.5mol,465.8g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00534mol,0.92g)和tbab(0.00071mol,0.23g),加热混合物并且抽真空至残压25mmhg,在108℃下回流10小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.2%,继续反应1.5小时,残余葡萄糖为0.08%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在118~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.76%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷溶液0.2
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
醇烷基糖苷溶液。测定其中的多糖的含量9.4%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.42。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为6.4:1。
[0074]
对比例4同实施例8相比,只采用了磺酸类化合物a组分和相转移催化剂中b1组分作为催化剂,其他同实施例8,反应转化为烷基糖苷的选择性变差,说明对比例4不能取得本技术的技术效果。
[0075]
对比例5
[0076]2‑
丙基庚醇烷基糖苷的合成
[0077]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.0mol,316.4g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00377mol,0.65g)和tbab(0.00124mol,0.40g)的复合催化剂,加热混合物并且抽真空至残压35mmhg,在108℃下回流10小时,当混合物变为半透明到透明状后监测反应至残余葡萄糖为0.06%,加入2.0g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度至112~178℃左右,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.27%,加去离子水溶解,配成固含量70%的水溶液,加入相对于烷基糖苷有效质量0.15
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量7.9%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.48。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为7.9:1。
[0078]
对比例5同实施例4相比,只采用了磺酸类化合物a组分和相转移催化剂中b1组分作为催化剂,其他同实施例4,反应略变慢,且反应转化为烷基糖苷的选择性变差,说明对比例5不能取得本技术的技术效果。
[0079]
对比例6
[0080]2‑
丙基庚醇烷基糖苷的合成
[0081]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(2.0mol,316.4g),无水葡萄糖(0.5mol,90g),在搅拌下加入bsa(0.005mol,0.81g)和tebab(0.00147mol,0.40g)的复合催化剂,加热混合物并且抽真空至残压25mmhg,在105℃下回流
10小时,当混合物变为半透明到透明状后监测反应至残余葡萄糖为0.22%,继续反应2小时残余葡萄糖0.12%,加入2.0g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度至108~175℃左右,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.31%,加去离子水溶解,配成固含量60%的水溶液,加入相对于烷基糖苷有效质量0.15
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量10.2%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.44。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为4.9:1。
[0082]
对比例6同实施例3相比,只采用了磺酸类化合物a组分和相转移催化剂中b1组分作为催化剂,其他同实施例3,反应略变慢,且反应转化为烷基糖苷的选择性变差,说明对比例6不能取得本技术的技术效果。
[0083]
对比例7
[0084]2‑
丙基庚醇烷基糖苷的合成
[0085]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丙基庚醇(1.5mol,237.3g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00377mol,0.65g)和tebac(0.00058mol,0.16g)的复合催化剂,加热混合物并且抽真空至残压35mmhg,在108℃下回流7.5小时,当混合物变为半透明到透明状后监测反应至残余葡萄糖为0.22%,继续反应2小时,残余葡萄糖为0.06%,加入2.2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在106~176℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.30%,加去离子水溶解,配成固含量60%的水溶液,加入相对于烷基糖苷有效质量0.2
‰
的mgo和2%的双氧水进行脱色,即得到淡黄色的2
‑
丙基庚醇烷基糖苷溶液。测定其中的多糖的含量8.7%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.46。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为5.9:1。
[0086]
对比例7同实施例2相比,只采用了磺酸类化合物a组分和相转移催化剂中b2组分作为催化剂,其他同实施例3,所得结果表明,反应变慢,且转化为烷基糖苷的选择性变差,说明对比例7不能取得本技术的技术效果。
[0087]
对比例8
[0088]2‑
丁基辛醇烷基糖苷的合成
[0089]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(1.5mol,279.5g),无水葡萄糖(0.5mol,90g),在搅拌下加入ptsa(0.00215mol,0.37g)和tebab(0.00272mol,0.74g),加热混合物并且抽真空至残压30mmhg,在115℃下回流7小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为1.09%,再延长时间3小时,监测反应至残余葡萄糖为0.23%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在120~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.64%,加去离子水溶解,配成固含量60%的水溶液,加入相对于烷基糖苷有效质量0.3
‰
的mgo和2%的双氧水进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液。测定其中的多糖的含量10.5%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.54。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多
糖的比例为4.7:1。
[0090]
对比例8同实施例11相比,只采用了磺酸类化合物a组分和相转移催化剂中b2组分作为催化剂,其他同实施例11,所得结果反应时间变长,且生成的多糖含量大,即反应转化为烷基糖苷的选择性变差,说明对比例8不能取得本技术的技术效果。
[0091]
对比例9
[0092]2‑
丁基辛醇烷基糖苷的合成
[0093]
在装有机械搅拌器,温度计,分水装置的四口烧瓶中加入2
‑
丁基辛醇(2.0mol,372.6g),无水葡萄糖(0.5mol,90g),在搅拌下加入bsa(0.00534mol,0.92g)和tebac(0.00202mol,0.46g),加热混合物并且抽真空至残压25mmhg,在108℃下回流11小时,当混合物由变为半透明到透明状后监测反应至残余葡萄糖为0.72%,继续回流2小时,监测反应至残余葡萄糖为0.15%,加入2g naoh溶液(32%)中和,然后将混合液转移至蒸馏装置脱醇,将压力调至残压为20mmhg,升高温度,在121~180℃蒸馏,至气相温度降至35℃以下时脱醇结束,得到褐色粘稠物,测残余醇为0.57%,加去离子水溶解,配成固含量60%的水溶液,加入相对于烷基糖苷有效质量0.2
‰
的mgo和2%的naclo进行脱色,即得到橙色的2
‑
丁基辛醇烷基糖苷溶液。测定其中的多糖的含量8.2%,根据gb/t 19464
‑
2004,测得烷基糖苷产品的平均聚合度为1.47。特殊支链伯醇烷基糖苷溶液中,特殊支链伯醇烷基糖苷与多糖的比例为6.3:1。
[0094]
对比例9同实施例10相比,只采用了磺酸类化合物a组分和相转移催化剂中b2组分作为催化剂,其他同实施例10,所得结果反应时间变长,且生成的多糖含量大,即反应的选择性变差,说明对比例9不能取得本技术的技术效果。
[0095]
由上述实施例可知,根据本发明提供的特殊支链伯醇合成烷基糖苷的制备方法,采用有机酸/二元复合相转移催化剂,有效提高了特殊支链伯醇尤其是特殊支链伯醇与葡萄糖缩醛反应活性,该方法工艺流程简洁,糖苷转化率高且副反应少,其中实施例1、3和4的产物中2
‑
丙基庚醇烷基糖苷与多糖的比例高达70:1以上,实施例8、10和11的产物中2
‑
丁基辛醇烷基糖苷与多糖的比例高达65:1以上。分别远远高于采用磺酸类化合物作为催化剂的对比例1、2以及采用磺酸类化合物与单一b(b1或b2)组合催化剂的3~7。其中实施例3和实施例4的效果最好。
[0096]
效果实施例
[0097]
本发明实施例制备的特殊支链伯醇烷基糖苷,反应体系中和后所得的混合液中特殊支链伯醇烷基糖苷含量高,具有表面张力低、润湿性性能优异、低泡等特点,实施例3制备的2
‑
丙基庚醇烷基糖苷和实施例4制备的2
‑
丁基辛醇烷基糖苷的红外光谱(ft
‑
ir)、液质联用(lc
‑
ms)的数据如下:
[0098]2‑
丙基庚醇烷基糖苷的红外光谱(ft
‑
ir):
as
(oh)3366.18cm
‑1,
as
(ch)2955.34cm
‑1,
s
(ch)2925.20cm
‑1,
s
(ch)2871.27cm
‑1,(c
‑
h)1456.81cm
‑1,1377.32cm
‑1,
as
(c
‑
o
‑
c)1150.09cm
‑1,1022.81cm
‑1;液质联用(lc
‑
ms):ms(m/z),365(m+hcoo
‑
),527(m+hcoo
‑
),689(m+hcoo
‑
)。
[0099]2‑
丁基辛醇烷基糖苷的红外光谱(ft
‑
ir):
as
(oh)3312.19cm
‑1,
as
(ch)2955.62cm
‑1,
s
(ch)2922.72cm
‑1,
s
(ch)2855.58cm
‑1,(c
‑
h)1466.11cm
‑1,1377.48cm
‑1,
as
(c
‑
o
‑
c)1145.79cm
‑1,1021.73cm
‑1,chch2‑
)724.48cm
‑1;液质联用(lc
‑
ms):m/z347(m
‑
1)393(m+
coo
‑
);m/z 509(m
‑
1),555(m+coo
‑
);1hnmr(400mhz,cdcl3)δ:0.86(2.0,m),1.26(16,m),1.62(1,m),3.20
‑
3.84(7.5,m),4.80
‑
5.20(1.8,m)。
[0100]
对实施例3制备的2
‑
丙基庚醇烷基糖苷和实施例4制备的2
‑
丁基辛醇烷基糖苷进行性能的测试,结果见表1所示:
[0101]
表1
[0102][0103]2‑
丙基庚醇烷基糖苷和2
‑
丁基辛醇烷基糖苷润湿性能均较好,具有较好的乳化性能,且泡沫较低。
[0104]
对实施例3制备的2
‑
丙基庚醇烷基糖苷和实施例4制备的2
‑
丁基辛醇烷基糖苷进行去油性能测试:
[0105]
上述产品配制质量分数为0.5%的水溶液,机油涂于不锈钢60mm*80mm*0.15mm试片上表面后常温放置10天,测定其对机油的去除性能,超声波震荡5min,分别在室温(25℃)和加热条件(40℃)下测试,结果如表2所示:
[0106]
表2
[0107][0108]
由表1可知,通过本发明的制备方法制备的特殊支链伯醇烷基糖苷,具有表面张力低、润湿和乳化性能优异、低泡等特点。由表2可知,在常温和加热条件下的去油性能均较佳,在常温下,实施例3制备的2
‑
丙基庚醇烷基糖苷和实施例4制备的2
‑
丁基辛醇烷基糖苷与传统的aeo9较为接近,而在加热条件下两者的去油较为彻底,优于aeo9。而通常工业生产中对清洗剂的要求是不同酸碱性条件下,要有良好的润湿、低泡性、去污能力强等性能,这些特殊性能是本发明制备的特殊支链伯醇烷基糖苷具有的,因而将具有较好的工业应用前景。
[0109]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1