一种倍半萜类化合物及其制备方法与流程
1.本发明涉及医药技术领域,尤其涉及一种倍半萜类化合物及其制备方法。
背景技术:
2.莪术为姜科姜黄属多年生草本植物蓬莪术curcuma phaeocaulis val.、广西莪术curcuma kwangsiensis s.g.lee et c.f.liang或温郁金curcuma wenyujin y.h.chen et c.ling的干燥根茎,栽培或野生于林荫下,分布于印度、马来西亚和中国各地。莪术性辛、苦,温,归肝、脾经。能行气破血,消积止痛。主治气血凝滞,心腹胀痛,症瘕,积聚,宿食不消,妇女血瘀经闭,跌打损伤作痛。
3.莪术根茎含挥发油,油中主成分为莪术呋喃烯酮、龙脑、大牻牛儿酮、α
‑
和β
‑
蒎烯、樟烯、柠檬烯、8
‑
按叶素、松油烯、异龙脑、丁香烯、姜黄烯、丁香烯环氧化物、姜黄酮、芳姜黄酮、莪术二酮、莪术烯醇、异莪术烯醇等。又含抗氧化剂活性的姜黄素类化合物。莪术油具有抗肿瘤、抗炎、抗菌、抗病毒、抗血栓等活性,其栓剂临床用于治疗白色念珠菌阴道感染、宫颈糜烂等。
技术实现要素:
4.有鉴于此,本发明要解决的技术问题在于提供一种倍半萜类化合物及其制备方法,由莪术油中分离得到一种倍半萜类化合物。
5.本发明提供了一种倍半萜类化合物,具有式i所示结构:
[0006][0007]
其化学名称为(4s,5s)
‑
牻牛儿酮
‑
4,5
‑
环氧化物,分子式为c
15
h
22
o2。
[0008]
本发明提供了上述倍半萜类化合物的制备方法,包括以下步骤:
[0009]
s1)将莪术的co2超临界萃取油进行第一次柱色谱分离,采用石油醚
‑
乙酸乙酯体系进行梯度洗脱,得到组分frg1~frg16;
[0010]
s2)取组分frg 8进行第二次柱色谱分离,采用甲醇
‑
水体系进行梯度洗脱,得到组分frg 8
‑
1~frg8
‑
47;
[0011]
s3)取组分frg 8
‑
4~frg 8
‑
10,进行第三次柱色谱分离,采用石油醚
‑
乙酸乙酯体系进行梯度洗脱,得到组分frg 8
‑4‑
1~frg 8
‑4‑
12;
[0012]
s4)取组分frg 8
‑4‑
5,进行重结晶,得到式i所示的倍半萜类化合物。
[0013]
本发明中,所述莪术的co2超临界萃取油为海南碧凯药业自制,优选的,其制备方法为鲜莪术块根烘干粉碎后,用超临界co2流体萃取器进行萃取。
[0014]
所述萃取的压力优选为9
‑
20mpa,萃取的温度优选为30
‑
60℃,萃取的时间优选为1.0
‑
3.0h。
[0015]
本发明优选的,所述第一次柱色谱分离中,石油醚和乙酸乙酯的体积比依次为100:0、100:5、100:10、100:20、100:30、0:100。
[0016]
本发明优选的,所述第一次柱色谱分离采用硅胶柱色谱。
[0017]
本发明优选的,所述第二次柱色谱分离中,甲醇和水的体积比依次为50:50、60:40、70:30、85:15、100:0。
[0018]
本发明优选的,所述第二次柱色谱分离采用rp
‑
18硅胶柱色谱。
[0019]
本发明优选的,所述第三次柱色谱分离中,石油醚和乙酸乙酯的体积比依次为100:0、100:3、100:6、100:9、100:12、0:100。
[0020]
本发明优选的,所述第三次柱色谱分离采用硅胶柱色谱。
[0021]
本发明优选的,所述重结晶的溶剂为chcl2‑
meoh体系。
[0022]
本发明优选的,所述chcl2和meoh的体积比为2:1。
[0023]
本发明中,在分离纯化时,用正相硅胶柱分离纯化时,优选采用200
‑
300目硅胶;用反相硅胶柱分离纯化时,优选采用aag12s50硅胶。
[0024]
本发明中,在采用硅胶薄层层析的方法检测式i所示化合物时,采用的显色剂为体积分数为1%的香草醛硫酸乙醇溶液,加热显色。
[0025]
通过结构检测,本发明制备得到的化合物为无色针状结晶(chcl2‑
meoh重结晶)。紫外254nm下观察呈暗斑,365nm下无荧光,10%硫酸乙醇试剂显色呈橙黄色,1%香草醛硫酸试剂显色呈紫红色斑点。[α]
d20
+261.3
°
(c 0.05,ch3oh)。uv(meoh)光谱显示最大吸收峰为λ
max
(logε)nm:313(2.77),239(3.55),205(3.78)。ir(kbr)光谱显示1678cm
‑1的共轭羰基伸缩振动吸收和1659cm
‑1的c=c双键伸缩振动吸收。esi
‑
ms给出准分子离子峰:m/z 235.3[m+h]
+
,257.3[m+na]
+
,491.1[2m+na]
+
,即分子量为234,结合氢谱和碳谱数据确定分子式为c
15
h
22
o2,提示为倍半萜类化合物。
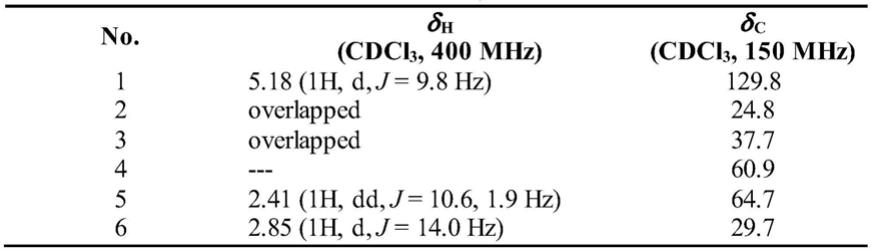
[0026]1h nmr(400mhz,cdcl3)(见表1)给出22个氢信号,其中4个甲基信号δ
h
1.80,1.79,1.70,1.01(各3h,s),1个烯氢信号δ
h 5.18(1h,d,j=9.8hz,h
‑
1)。
13
c nmr(150mhz,cdcl3)(见表1)给出15个碳信号,其中1个酮羰基信号δ
c 204.8,2组烯碳信号δ
c 134.8(c
‑
11),133.8(c
‑
10),129.8(c
‑
1),126.7(c
‑
7),以及1个三元氧环上的两个连氧碳信号δ
c 64.7(c
‑
5),60.9(c
‑
4)。根据以上信息,推测该化合物为牻牛儿酮的4,5
‑
环氧化物。将其波谱数据与文献[刘晓宇,楼燕,胡丹,陈丽霞,卜光明,邱峰.温郁金挥发油的化学成分.沈阳药科大学学报,2007,24(11):682
‑
686;kuroyanagi m,ueno a,ujiie k,sato s.structures of sesquiterpenes from curcuma aromatica salisb..chemical&pharmaceutical bulletin,1987,35(1):53
‑
59]对照,与牻牛儿酮
‑
4,5
‑
环氧化物的数据基本一致,故确定该化合物为牻牛儿酮
‑
4,5
‑
环氧化物。为了确定其绝对构型,测试了圆二色谱(ecd),该化合物的ecd(ch3oh):nm(δε):310(+4.61),254(+4.58),228(
‑
4.02),209(+21.90),与文献[yoshihara m,shibuya h,kitano e,yanagi k,kitagawa i.the absolute stereostructure of(4s,5s)
‑
germacrone
‑
4,5
‑
epoxide from zedoariae rhizoma cultivated in yakushima island.chemical&pharmaceutical bulletin,1984,32(5):2059
‑
2062]报道的(4s,5s)
‑
牻牛儿酮
‑
4,5
‑
环氧化物的ecd谱一致,故确定化合物为(4s,
5s)
‑
牻牛儿酮
‑
4,5
‑
环氧化物[(4s,5s)
‑
germacrone
‑
4,5
‑
epoxide]。1h
‑
nmr和
13
c
‑
nmr数据见表1。
[0027]
表1c15h22o2(in cdcl3)的nmr波谱数据
[0028][0029][0030]
与现有技术相比,本发明提供了一种倍半萜类化合物,具有式i所示结构。上述化合物的成功提取为开发新药提供了化学实体或先导化合物。
附图说明
[0031]
图1为本发明制备的式i所示化合物的核磁氢谱图。
具体实施方式
[0032]
为了进一步说明本发明,下面结合实施例对本发明提供的倍半萜类化合物及其制备方法进行详细描述。
[0033]
以下实施例中的莪术co2超临界萃取油按照以下方法制备:鲜莪术块根经称量清洗挑选后,切成厚度≦1.0cm的片状,烘近干后粉碎,用超临界co2流体萃取器进行萃取,得到萃取油。萃取的压力为9
‑
20mpa,萃取的温度为30
‑
60℃,萃取的时间为1.0
‑
3.0h。
[0034]
实施例1
[0035]
莪术co2超临界萃取油519.80g经硅胶柱色谱,用石油醚
‑
乙酸乙酯系统(100:0、100:5、100:10、100:20、100:30、0:100)进行梯度洗脱,每瓶500ml接收一个流份,经tlc检测,合并为16个部分,其中,第一部分3.51g,第二部分19.29g,第三部分90.20g,第四部分103.97g,第五部分22.06g,第六部分151.76g,第七部分61.73g,第八部分48.51g,第九部分6.92g,第十部分16.55g,第十一部分7.20g,第十二部分10.43g,第十三部分4.56g,第十四部分8.38g,第十五部分15.86g,第十六部分9.42g。
[0036]
粗分第八部分样品44.64g经rp
‑
18硅胶柱色谱,用甲醇
‑
水系统(50:50、60:40、70:
30、85:15、100:0)进行梯度洗脱,每瓶300ml接收一个流份,共收集47个流份。其中,流份4
‑
10(29.59g)经硅胶柱色谱,用石油醚
‑
乙酸乙酯系统(100:0、100:3、100:6、100:9、100:12、0:100)进行梯度洗脱,每瓶500ml接收一个流份,共收集118个流份,经tlc检测,合并为12个部分,其中,第一部分0.22g,第二部分0.03g,第三部分0.08g,第四部分0.74g,第五部分7.97g,第六部分0.30g,第七部分0.63g,第八部分5.82g,第九部分3.56g,第十部分1.47g,第十一部分0.24g,第十二部分8.95g。
[0037]
第五部分样品经chcl2‑
meoh重结晶,得到式i所示化合物,命名为(4s,5s)
‑
牻牛儿酮
‑
4,5
‑
环氧化物,分子式为c
15
h
22
o2。
[0038]
实施例2结构鉴定
[0039]
利用波谱技术,包括紫外、红外、核磁共振、质谱、圆二色谱测试,对实施例1中得到的化合物进行鉴定;其波谱数据如下:
[0040]
无色针状结晶(chcl2‑
meoh)。
[0041]
[α]
d20
+261.3
°
(c 0.05,ch3oh)。
[0042]
uv(meoh)λ
max
(logε)nm:313(2.77),239(3.55),205(3.78)。
[0043]
ecd(ch3oh):nm(δε):310(+4.61),254(+4.58),228(
‑
4.02),209(+21.90)。
[0044]
ir(kbr):2979,2926,2862,1678,1659,1436,1386,1253cm
‑1。
[0045]
esi
‑
ms:m/z 235.3[m+h]
+
,257.3[m+na]
+
,491.1[2m+na]
+
。
[0046]1h nmr(400mhz,cdcl3)和
13
c nmr(150mhz,cdcl3)数据见表1。
[0047]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1