一种氯舒隆的合成方法与流程
1.本发明涉及有机合成技术领域,尤其涉及一种氯舒隆的合成方法。
背景技术:
2.氯舒隆别名克洛索隆,氯索龙,分子式:c8h8cl3n3o4s2,为苯磺酰胺类化合物,是一种抗血虫药,用于血虫病的治疗。
3.氯舒隆的化学结构式如下:
[0004][0005]
cn104557623a公开了一种4
‑
氨基
‑6‑
(三氯乙烯基)
‑
1,3
‑
苯二磺胺的制备方法,其中还原反应中采用铱络合物催化剂,铱络合物催化剂价格昂贵,不利于工业化生产。
[0006]
cn104230767a公开了一种氯舒隆的制备方法,其中还原反应采用水合肼,同时以三氯化铁为催化剂,以活性炭为催化剂载体。其反应速度较慢,催化还原的效果不理想,并且后处理过程中抽滤速率较慢,产生的碳泥只能作为危废处理,不利于工业化生产。
[0007]
鉴于现有氯舒隆的制备方法还存在各种缺陷,特提出本发明。
技术实现要素:
[0008]
本发明的目的在于提供一种氯舒隆的合成方法。
[0009]
具体地,本发明提供如下技术方案:
[0010]
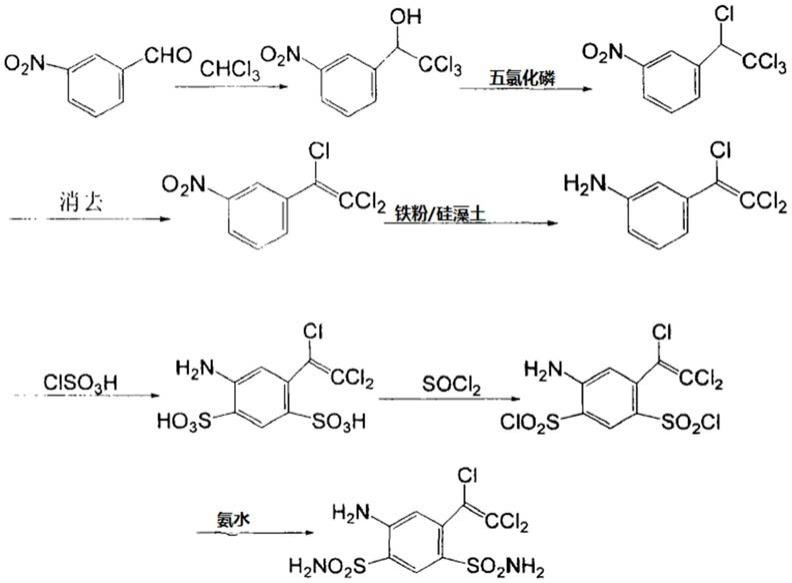
一种氯舒隆的合成方法,以间硝基苯甲醛和三氯甲烷为初始原料,依次经缩合反应、氯代反应、消去反应、还原反应、氯磺化反应、胺化反应制得氯舒隆;其中,所述还原反应在含有盐酸、铁粉和硅藻土的体系中进行。
[0011]
本发明对氯舒隆的合成方法,特别是还原反应进行了大量的研究,发现在铁粉/盐酸的还原体系中加入硅藻土,在保证收率的同时,加快3
‑
三氯乙烯基硝基苯的还原速率,缩短反应时间,反应副产物氢气释放缓慢,提高反应安全性,以及简化后处理过程,适合于工业化生产。
[0012]
合成路线如下:
[0013][0014]
在本发明一个优选实施方式中,所述铁粉和硅藻土的重量比为1:(1.0
‑
1.2)。
[0015]
进一步地,所述铁粉的粒径为200
‑
300目。
[0016]
进一步地,所述盐酸的浓度为4
‑
8mol/l,铁粉与盐酸的摩尔体积比为1mol:(0.8
‑
0.9)l。
[0017]
在本发明一个优选实施方式中,所述还原反应在甲醇或乙醇存在下进行。
[0018]
进一步地,所述还原反应的温度为20
‑
25℃。
[0019]
进一步地,所述铁粉与消去反应所得产物3
‑
三氯乙烯基硝基苯的质量比为(0.8
‑
0.85):1。
[0020]
在本发明一个优选实施方式中,所述缩合反应在第一催化剂和第一溶剂存在下进行,间硝基苯甲醛、三氯甲烷、第一催化剂的摩尔比为1:(1.5
‑
1.6):(1
‑
1.12)。
[0021]
进一步地,所述第一催化剂选自六氢吡啶、四氢吡咯、哌啶中的任一种。
[0022]
进一步地,所述第一溶剂选自二氯甲烷、n,n
‑
二甲基甲酰胺、二甲基亚砜、二氯乙烷中的任一种。
[0023]
进一步地,所述缩合反应的温度为
‑5‑
0℃。
[0024]
在本发明一个优选实施方式中,所述氯代反应在氯化试剂和缚酸剂存在下进行,所述氯化试剂、缚酸剂和缩合反应所得产物三氯甲基
‑3‑
硝基卞醇的摩尔比为(0.5
‑
0.6):(1.1
‑
1.2):1。
[0025]
进一步地,所述氯化试剂选自氯化亚砜、三氯氧磷、五氯化磷中的任一种。
[0026]
进一步地,所述缚酸剂为吡啶或三乙胺。
[0027]
进一步地,所述氯代反应的温度为30℃以下。
[0028]
在本发明一个优选实施方式中,所述消去反应在第二催化剂和第二溶剂存在下进行,所述第二催化剂与氯代反应所得产物3
‑
四氯乙基硝基苯的摩尔比为(1.05
‑
1.1):1。
[0029]
进一步地,所述第二催化剂选自氢氧化钠、氢氧化钾、乙醇钠中的任一种。
[0030]
进一步地,所述第二溶剂为甲醇或乙醇。
[0031]
进一步地,所述消去反应的温度为25
‑
40℃。
[0032]
在本发明一个优选实施方式中,所述氯磺化反应使用的磺化试剂为氯磺酸或浓硫酸;使用的氯化试剂为氯化亚砜。
[0033]
进一步地,所述氯磺化反应的温度为100
‑
130℃。
[0034]
在本发明一个优选实施方式中,所述胺化反应使用的胺化试剂为氨气或氨水。
[0035]
进一步地,所述胺化反应的温度为20
‑
25℃。
[0036]
在本发明一个优选实施方式中,所述方法还包括对胺化反应的产物进行纯化,所述纯化包括降温结晶和重结晶。
[0037]
进一步地,所述降温结晶采用的溶媒为乙酸乙酯、二氯甲烷、正庚烷、正己烷中的一种或几种;所述重结晶采用的溶媒为甲醇、乙醇、水中的一种或几种。
[0038]
本发明的有益效果在于:
[0039]
本发明方法所获得的产品质量稳定,收率高,并且工艺相对简单,可操作性强,安全系数高,具备良好的工业化前景。
具体实施方式
[0040]
以下实施例用于说明本发明,但不用来限制本发明的范围。
[0041]
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
[0042]
实施例1
[0043]
本实施例提供一种氯舒隆的合成方法,包括以下步骤:
[0044]
(1)缩合反应
[0045]
1、准备圆底玻璃烧瓶一个,在烧瓶中加入30克的对硝基苯甲醛,量取45毫升的三氯甲烷和120毫升的dmf,放入磁子搅拌溶解,开启冰机设定温度
‑
5℃,将上述混合体系降温至
‑
5℃
‑
0℃,备用。
[0046]
2、量取15毫升的六氢吡啶于烧杯中,加入30毫升的甲醇混合均匀后转入滴液漏斗内,备用。
[0047]
3、搅拌下向1.1所得混合体系中缓慢滴加1.2所得六氢吡啶的甲醇溶液,滴加完毕保持低温(
‑
5℃)30min以上,搅拌下加入一定量的稀盐酸中和反应,ph值监控接近中性下加入150毫升的甲苯,继续搅拌两个小时,取出烧瓶,升温至25℃,用分液漏斗收集甲苯层(上层)。有机相用水洗涤两次,去除甲醇和盐分以及dmf等残留。
[0048]
4、向1.3所得有机相中加入20克活性炭,升温至40℃搅拌30min以上,抽滤。有机相用5%的碳酸钠洗涤一次后用纯化水洗涤一次。
[0049]
5、有机相减压浓缩至干,加入正己烷30毫升,溶清后降温至
‑
2℃,抽滤得到晶体,正己烷洗涤后干燥,得到三氯甲基
‑3‑
硝基苄醇,收率91.5%,hplc 98.9%。
[0050]
(2)氯代反应
[0051]
1、在1000毫升的三口瓶中,装置温度计和机械搅拌,另一口加分液漏斗,烧瓶内加入30克的五氯化磷和90毫升的二氯甲烷,搅拌下溶解,如果溶解不完可以加热溶解完全。备
用。
[0052]
2、将30克的三氯甲烷
‑3‑
硝基苄醇溶解于90毫升的二氯甲烷中,加热下溶解完全,转入滴液漏斗中,备用。
[0053]
3、将2.2所得溶液缓慢滴加到2.1的三口瓶中,滴加时间约1小时,滴加完毕控制反应体系的温度在30℃以下,并搅拌3小时以上,通过tlc判断反应的完全程度,反应原点消失证明反应完毕。
[0054]
4、反应完毕后加入小冰渣终止反应,并且剧烈搅拌,待无氯化氢气体放出停止加入冰渣,继续搅拌1小时。静置分层收集有机相,水相加入二分之一体积的二氯甲烷再次萃取,静止分层收集有机相,合并两次的有机相。先用饱和食盐水洗涤,再用纯化水洗涤,然后用氢氧化钠溶液(饱和)洗涤,中和有机相中残留的氯化氢,最后用纯化水洗涤,洗到ph值接近中性为止。合并有机相再用水洗涤,减压浓缩至干为金黄色油状液体,降温后为无色冰糖装晶体。干燥备用,得到3
‑
四氯乙基硝基苯,收率98.1%,hplc 99.2%。
[0055]
(3)消去反应
[0056]
1、常温下将30克的氢氧化钠溶解到100毫升的甲醇中,溶清后导入1000毫升的三口瓶中,加装机械搅拌和温度计,备用。
[0057]
2、将30克的3
‑
四氯乙基硝基苯溶解于150毫升的甲醇中,溶清后导入滴液漏斗中,控制滴加速度缓慢滴加,1个小时滴加完毕。
[0058]
3、反应过程中体系的温度会缓慢的升高,可不做处理,反应过程中有白色的固体析出。滴加结束继续搅拌2个小时左右,加入浓盐酸中和氢氧化钠,ph接近中性即可。过滤,水洗涤,干燥,得到产物三氯乙烯基硝基苯,收率89.2%,hplc 97.2%。
[0059]
(4)还原反应
[0060]
1、将30克的三氯乙烯基硝基苯和27克的还原铁粉以及30克的硅藻土于圆底烧瓶中,加入三氯乙烯基硝基苯150毫升(m/v)的50%乙醇水溶液(v/v),机械搅拌下升温至50℃左右。
[0061]
2、在滴液漏斗中加入4m的盐酸40毫升,并且加入40毫升的50%乙醇水溶液,边搅拌边滴加。
[0062]
3、滴加完毕后升温至95
‑
100℃,回流1.5
‑
2小时,趁热减压过滤,固形物用50%的乙醇水溶液洗涤三次,收集滤液。为了提高收率,滤饼可以用热乙醇洗涤一次,合并所有的乙醇溶液,用饱和碳酸钠溶液中和,至ph为中性。
[0063]
4、乙醇水溶液用0.5bv二氯甲烷萃取,二氯甲烷溶液用饱和食盐水洗涤三次,然后用无水硫酸钠干燥,得到产物3
‑
三氯乙烯基苯胺,收率89.5%,hplc 93.5%,作为反应中间体备用。
[0064]
(5)氯磺化反应
[0065]
1、上述脱水后的3
‑
三氯乙烯基苯胺的二氯甲烷溶液中滴加氯磺酸130毫升,温度控制在10℃以内,滴加时间5min。
[0066]
2、滴加完毕后可以观察到有胶状粘性物质生成,搅拌下升温至130℃,搅拌两个小时后反应体系为黑色焦糖状。
[0067]
3、tlc跟踪3
‑
三氯乙烯基苯胺的原点消失后,滴加70毫升的二氯亚砜,滴加完毕加热至回流2小时。反应过程中有大量的气泡产生。
[0068]
4、冷却后有产品析出,将反应体系中加入二氯甲烷的冰溶液,搅拌下抽滤,得到固体,水洗干燥,得到4
‑
氨基
‑6‑
三氯乙烯基
‑
1,3
‑
苯二磺酰氯,收率75.6%,hplc 91.5%。
[0069]
(6)胺化反应
[0070]
1、将30克的4
‑
氨基
‑6‑
三氯乙烯基
‑
1,3
‑
苯二磺酰氯溶解于60毫升二氯甲烷中,搅拌下加入240毫升的氨水,持续搅拌12小时以上,
[0071]
2、调节ph至10左右,产品大量析出,抽滤得到固体产品,粗干燥。
[0072]
3、粗品用乙酸乙酯溶解后加入三倍体积的纯化水,搅拌后收集有机相,水相用一定量的乙酸乙酯萃取,合并有机相,乙酸乙酯相用10%的食盐水洗涤,无水硫酸钠干燥,减压浓缩至干。
[0073]
4、加入正己烷热熔,产品成泥浆状,然后加入20:7的乙酸乙酯和正己烷下冰浴2小时,抽滤得到产品。干燥得到白色固体。
[0074]
5、甲醇和水重结晶,成品干燥的温度为60℃
‑
80℃真空干燥,总收率54.2%,hplc 99.8%。
[0075]
实施例2
[0076]
本实施例提供一种氯舒隆的合成方法,其与实施例1的区别仅在于,缩合反应中:量取15毫升的哌啶烧杯中,加入30毫升的乙醇混合均匀后转入滴液漏斗内,备用。缩合反应所得产物三氯甲基
‑3‑
硝基苄醇的收率90.1%,hplc 98.4%。
[0077]
对比例1
[0078]
本对比例提供一种氯舒隆的合成方法,其与实施例1的区别仅在于还原反应不同。本对比例未添加硅藻土。还原反应所得产物3
‑
三氯乙烯基苯胺的收率81.5%,hplc 91.2%。
[0079]
对比例2
[0080]
本对比例提供一种氯舒隆的合成方法,其与实施例1的区别仅在于还原反应不同。本对比例用等量的活性炭替换硅藻土。在趁热减压过滤步骤中发现过滤困难、滤速较慢。还原反应所得产物3
‑
三氯乙烯基苯胺的收率82.7%,hplc 91.5%。活性炭代替硅藻土的收率和纯度低于硅藻土,初步分析活性炭对产品造成死吸附,导致收率偏低。
[0081]
对比例3
[0082]
本对比例提供一种氯舒隆的合成方法,其与实施例1的区别仅在于还原反应不同。本对比例添加的硅藻土的量为15克。还原反应所得到产物3
‑
三氯乙烯基苯胺的收率83.9%,hplc 91.9%。
[0083]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1