一种氨基脂质及其应用
1.本发明医药化学技术领域,特别涉及一种氨基脂质及其应用。
背景技术:
2.基因药物是指将外源正常基因导入靶细胞,以纠正或补偿因基因缺陷和异常引起的疾病,以达到治疗目的;或者基因得到表达,产生相应的抗原,进而诱导记忆性的免疫反应。在过去的二十多年间,基因药物在很多疾病治疗领域将研究从临床前推向了临床,对于由基因异常引起的,医药界至今难以解决的疾病如肿瘤等具有无可替代的优势。常见的基因药物有质粒dna (plasmid dna ,pdna) 、反义寡核昔酸(antisense odn) 、小干扰rna (sirna) 和信使rna (mrna) 。
3.然而,将外源基因引入体内,其会被体内的核酸酶降解,在未进入靶细胞之前,便被降解成小分子核苷酸,从而失去治疗作用。因此,实现基因治疗的关键是高效、安全的基因递送系统。基因载体在运送基因的时候要经历多个复杂的过程:通过血液循环到达靶细胞,细胞摄取,内涵体的逃逸,胞内运动,载体释放基因物质。其主要障碍主要是复杂血液环境的细胞外障碍和溶酶体酶降解的细胞内障碍。因此寻找良好的基因载体,使得靶基因到达靶点发挥效用,是基因载体研究者亟待解决的问题。
4.目前,在基因输送载体系统方面主要分为两大类:一是病毒载体系统,二是非病毒载体系统。病毒载体是一种天然的载体资源,病毒基因组结构简单,转染效率高,靶细胞特异性强,但其导向性差、携带能力低、免疫原性等缺点限制了其使用。因此多样性、无免疫原性及易于控制生产的非病毒载体系统近年来备受关注,并在很多治疗领域有所应用。常用的非病毒载体系统主要是阳离子脂质(cationic lipids) 载体。
5.阳离子脂质有三个重要的结构区域:带正电荷的亲水极性头部基因,中间负责连接极性和非极性的两端的连接链和疏水脂质链。含胺类基团的极性头部起着脂质体与rna,脂质体/rna 复合物与细胞膜相互结合的作用,影响脂质带电情况,在溶酶体逃逸过程起主要作用。连接链决定了阳离子脂质体的化学和生物稳定性,特别是因此而产生的细胞毒性。疏水区可以为碳链形式或类固醇等多种结构,并且碳链的长度、是否饱和和具体类型将影响脂质行为,其既为脂双层提供足够的流动性,又能促使阳离子脂质体在体内的脂质融合。
6.阳离子脂质体与带负电的基团通过静电作用形成脂质体/基因复合物。复合物因阳离子脂质体的过剩带正电,带正电的脂质体/基因复合物由于静电作用吸附于带负电的细胞表面。然后通过与细胞膜融合或细胞的内吞作用进入细胞内。阳离子脂质用于基因治疗的主要特点是在核内体逃逸过程中的电荷影响的膜融合作用。但同时,阳离子脂质/基因复合物过剩的正电以及部分阳离子脂质难降解的特性也导致了细胞毒性。因此较低的转染效率和细胞毒性是限制阳离子脂质应用的主要缺点。目前阳离子脂质作为基因载体因其结构简单、操作简便、生物安全性高等特点成为了目前应用最为广泛的非病毒载体,但其转染的效率低、正电荷所导致的细胞毒性问题仍待解决,因此本发明尝试设计可电离阳离子脂质来解决上述问题,以达到较好的基因治疗效果。
技术实现要素:
7.针对现有技术中阳离子脂质体的转染的效率低、正电荷所导致的细胞毒性等技术问题,本发明提供了一种氨基脂质及其应用。
8.本发明的目的通过以下技术方案予以实现:一种氨基脂质,其结构如式(i)所示:其中,l为c1‑
c
24
亚烷基、c1‑
c
24
亚烯基、c3‑
c8亚环烷基、c3‑
c8亚环烯基;r1为h、or5、cn、
‑
c (=o) or4、
‑
oc(=o) r4、
‑ꢀ
c (=o) n r4r5、
‑
nr
5 c (=o) r4或n r4r5;r2、r3、r4和r5彼此相同或不同,并且各自独立地选自h, c1‑
c
24
烷基、c2‑
c
24
烯基、c2‑
c
24
炔基;所述c1‑
c
24
烷基、c2‑
c
24
烯基、c2‑
c
24
炔基可任选地被c1‑
c6烃基取代;或r2和r3相连接形成4~10元杂环,其中,多元杂环包含1~6个杂原子,所述杂原子选自氮、硫或氧。
9.优选地,所述r2选自c6‑
c
24
烷基、c6‑
c
24
烯基、c6‑
c
24
炔基;所述被c6‑
c
24
烷基、c6‑
c
24
烯基、c6‑
c
24
炔基可任选地被c1‑
c6烃基取代。
10.优选地,以nh2作为自由基的位置,l和r1相连形成nh2‑
l
‑
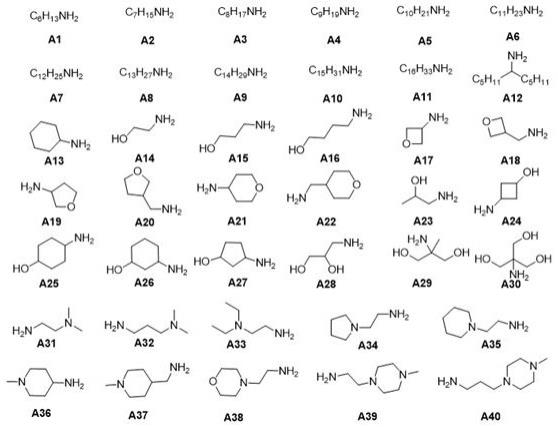
r1选自a1、a2、a3、a4、a5、a6、a7、a8、a9、a10、a11、a12、a13、a14、a15、a16、a17、a18、a19、a20、a21、a22、a23、a24、a25、a26、a27、a28、a29、a30、a31、a32、a33、a34、a35、a36、a37、a38、a39、a40中的一种。
11.在式(i)的化合物中l和r1相连以后与n原子连接。上述nh2‑
l
‑
r1中nh2取代的位置即为与式(i)化合物相接的自由基位置。
12.优选地,r2、r3与相邻的n原子形成r2r3‑
nh,其中h作为自由基的位置;r2r3‑
nh选自n1、n2、n3、n4、n5、n6、n7、n8、n9、n10、n11、n12、n13、 n14、 n15、n16、n17、 n18、 n19、n20、n21、 n22、n23中的一种。
13.在式(i)的化合物中r2、r3连接在同一n原子上,n原子再与磺酰基上的s原子连接。上述r2r3‑
nh中h原子即为与式(i)中s原子连接的位置。
14.所述氨基脂质的制备方法,包括以下步骤:s1. 化合物nh2‑
l
‑
r1与乙烯基磺酰氟在溶剂中搅拌反应;s2. 在步骤s1反应体系中加入r2r3nh,并在碱存在的条件下加热反应即得。
15.反应的流程如下:优选地,所述方法包括以下步骤:(1)在
‑
20℃至40℃的温度下,在乙烯基磺酰氟(esf)与由r1
‑
l
‑
nh2表示的化合物进行第一反应,得到第一中间体;(2)在分离或不分离所述第一中间体的情况下,在作为缚酸剂的碱的存在下,使所述第一中间体与由hnr2r3表示的胺在加热条件进行第二反应,得到所述式i的氨基脂质化合物。优选地,上述步骤s2中加热温度为50~120℃。上述制备方法中使用的碱为有机碱或无机碱,如:三乙胺、dipea、吡啶、dmap、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾等所述氨基脂质及其药物可接受的盐、前药或立体异构体在制备用于基因治疗、基因疫苗接种、反义治疗或通过干扰rna的治疗的药物中的应用。
16.优选地,上述应用为在制备用于癌症或遗传疾病的治疗药物中的应用。
17.优选地,上述应用为在制备用于肺癌、胃癌、肝癌、食管癌、结肠癌、胰腺癌、脑癌、淋巴癌、血癌或前列腺癌药物中的应用,所述遗传疾病为血友病,地中海贫血、高雪氏病中的一种或多种。
18.优选地,上述应用为在制备用于治疗癌症、过敏、毒性和病原体感染药物中的应
用。
19.优选地,上述应用为在制备用于核酸转移的药物中的应用。
20.优选地,所述核酸为rna、信使rna (mrna)、反义寡核苷酸、dna、质粒、核糖体rna(rrna)、微rna(mirna)、转移rna(trna)、小的抑制rna(sirna)和小的核rna(snrna)。
21.与现有技术相比本发明具有以下技术效果:本发明公开的一种氨基脂质化合物,通过乙烯基磺酰氟(esf)这一双功能的亲电试剂,将胺基头部基团和疏水链构建至氨基脂质中,充分利用了esf的点击化学反应特性,构建氨基脂质的过程中反应条件温和,不需要保护和脱保护,原子经济性高。在体外、体内的递送研究中,显示了优良的递送核酸至细胞中的能力。上述氨基脂质化合物具备两个磺酰胺,该基团的引入显著增强了脂质纳米颗粒的稳定性,改善了体内循环时间,从而提高了体内递送效率。所述氨基脂质化合物的制备方法具有原料易得、反应条件温和、反应选择性好、反应产率高、仪器设备要求低和操作简单的优点。
附图说明
22.图1实施例10中代表性氨基脂质化合物递送ova mrna刺激bmdc后,分化成呈递ova抗原的细胞群体的比例图;图2实施例10中代表性氨基脂质化合物递送ova mrna刺激bmdc后,分化为成熟dc细胞群体的比例图;图3实施例12中代表性氨基脂质化合物皮下给药递送ova mrna所产生的体液抗体滴度图;图4实施例13中肌肉注射ova mrna疫苗后的荷瘤小鼠生存曲线图。
具体实施方式
23.下面对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
24.下述实验例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
25.本发明所用的术语“任选地取代的”意指与原子或基团连接的一个或多个氢原子独立地未被取代,或被一个或多个例如一、二、三或四个取代基取代。当一个原子或基团被多个取代基取代时,所述多个取代基可以相同或不同。
26.文中的缩写:dna
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
脱氧核糖核酸rna
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
核糖核酸dope
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二油酰基磷脂酰乙醇胺dspc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二硬脂酰磷脂酰胆碱peg2000
‑
dmg
ꢀꢀ
(1
‑ꢀ
(单甲氧基聚乙二醇)
ꢀ‑
2, 3 二肉豆寇酰基甘油kd千道尔顿pbs
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
磷酸盐缓冲溶液。
27.实施例1 a1ny系列氨基胺基脂质化合物库的平行合成与表征在250ml的反应瓶中依次加入正已胺(25.3 mg, 0.25 mmol),乙烯基磺酰氟(55 mg, 0.5 mmol),无水四氢呋喃2.5 ml, 室温下搅拌反应5min,得step i反应液(2.5 ml, 0.1m)。
28.用移液枪将每个step i反应液分别转移至22个1.5 ml的ep管中(每个0.1ml, 0.01 mmol),相应的ep管中各加入二胺的thf溶液(0.12 ml, 0.024 mmol,0.2m)、dipea 的thf溶液(0.2 ml, 0.04 mmol, 0.2m),于加热型振摇反应器(thermo
‑
shaker)中于78℃反应1 h,tlc检测无step i 原料。反应结束后,将反应管内的溶剂常温挥干,即得到23个胺基脂质化合物a1ny。进行质谱检测,结果见下面的表1。
29.表1:a1ny系列氨基胺基脂质化合物库的mw/z值
80 mmol),二异丙基乙基胺(20.6 g, 160 mmol)升温至75 ℃反应4 h。浓缩后,使用快速柱层析系统纯化(二氯甲烷:甲醇= 20:1至5:1)得到化合物a12n2(5.06 g, 76%)。1h nmr (400 mhz, dmso
‑
d6):δ 3.62 (m, 4h), 3.03 (m, 2h), 2.94 (m, 4h), 2.89 (m, 4h), 1.54
‑
1.23 (m, 50h), 0.89 (m, 9h). esi
‑
ms calculated for c
35
h
76
n3o4s
2+ [m+h]
+ 667.1, found 667.3。
[0031]
实施例4 2,2'
‑
(壬基氮杂二基)双(n
‑
十一烷基乙烷
‑1‑
磺酰胺)在250ml的反应瓶中依次加入乙醇胺(0.61 g, 10 mmol),乙烯基磺酰氟(2.2 g, 20 mmol),无水四氢呋喃80 ml, 室温下搅拌反应5min,再加入正十五烷基甲基胺 (14.5 g, 60 mmol),二异丙基乙基胺(20.6 g, 160 mmol)升温至75 ℃反应4 h。浓缩后,使用快速柱层析系统纯化(二氯甲烷:甲醇= 20:1至5:1)得到化合物a14n13(5.06 g, 76%)。1h nmr (400 mhz, dmso
‑
d6):δ 3.62 (m, 4h), 2.49 (m, 1h), 2.94 (m, 4h), 2.89 (m, 4h), 1.54
‑
1.23 (m, 44h), 0.89 (m, 12h). esi
‑
ms calculated for c
33
h
72
n3o4s
2+ [m+h]
+ 667.1, found 667.3。
[0032]
实施例52,2'
‑
(壬基氮杂二基)双(n
‑
十一烷基乙烷
‑1‑
磺酰胺)在250ml的反应瓶中依次加入4
‑
氨基四氢吡喃(1.01 g, 10 mmol),乙烯基磺酰氟(2.2 g, 20 mmol),无水四氢呋喃80 ml, 室温下搅拌反应5min,再加入二已胺 (14.8 g, 80 mmol),二异丙基乙基胺(20.6 g, 160 mmol)升温至75 ℃反应4 h。浓缩后,使用快速柱层析系统纯化(二氯甲烷:甲醇= 20:1至5:1)得到化合物a21n16(4.43 g, 68%)。1h nmr (400 mhz, dmso
‑
d6):δ 3.69
‑
3.61 (m, 4h), 3.52 (m, 4h), 2.94 (m, 4h), 2.72 (m, 1h), 2.39 (m, 8h), 1.78
‑
1.52 (m, 4h), 1.39
‑
1.23 (m, 32h), 0.89 (m, 12h). esi
‑
ms calculated for c
33
h
70
n3o5s
2+ [m+h]
+ 652.5, found 652.9。
[0033]
实施例62,2'
‑
(壬基氮杂二基)双(n
‑
十一烷基乙烷
‑1‑
磺酰胺)在250ml的反应瓶中依次加入n, n
‑
二甲基丙二胺(204 mg, 2 mmol),乙烯基磺酰氟(440 mg, 4 mmol),无水四氢呋喃40 ml, 室温下搅拌反应5min,再加入甲基二烯十八烷
基胺(3.36g,12mmol),二异丙基乙基胺(4.12g,32mmol)升温至75℃反应4h。浓缩后,使用快速柱层析系统纯化(二氯甲烷:甲醇=20:1至5:1)得到化合物a32n23(1.41g,84%)。1hnmr(400mhz,dmso
‑
d6):δ5.42
‑
5.28(m,8h),3.52(m,4h),2.94(m,4h),2.89(s,6h),2.80(m,4h),2.39
‑
2.35(m,8h),2.16(m,8h),2.14(m,6h),1.54
‑
1.23(m,38h),0.89(m,6h).esi
‑
mscalculatedforc
47
h
93
n4o4s
2+
[m+h]
+
841.7,found841.9。
[0034]
实施例7氨基脂质化合物作为mrna载体的体外评价细胞系:hela细胞系(atcc)培养基:补充了10%胎牛血清的dmem(invitrogen)筛选形式:96孔板细胞转染检测(读出):相对于总细胞数(使用核染料hoechst测定总细胞数-参见图2)的gfp荧光细胞数百分比例。根据制造商的说明,lipofectamine2000(invitrogen)用作阳性对照组。
[0035]
方法:使用8通道移液管加样。所示的含量为96孔平板的单孔。
[0036]
1.将实施例1中所述的氨基脂质化合物与二油酰基磷脂酰乙醇胺(dope),胆固醇,peg2000
‑
dmg的摩尔比为45:10:42.5:2.5的比例混合溶解在无水乙醇中;egfpmrna(trilink)溶解在醋酸钠溶液(50mm,ph=4.0)中,使用排枪取出上述的混合脂质溶液,加入至egfp
‑
mrna溶液中,充分混合,控制其配比为乙醇溶液与醋酸钠溶液(50mm,ph=4.0)的比例为1:3,制得脂质纳米颗粒溶液。氨基脂质化合物与绿色荧光蛋白mrna(egfpmrna)的质量比约为8:1,每孔mrna的用量为100ng。
[0037]
2.脂质纳米颗粒溶液在室温下孵育30min后,加入90μl新鲜重悬浮的hela细胞(3
‑5×
104细胞),并用移液管混合。将100μl的细胞+脂质纳米颗粒转移至96
‑
孔培养平板的分开孔中,并且置于37℃的含有5%co2的培养箱中。
[0038]
3.细胞初始转染后20至24小时,将hoechst33258(invitrogen)以0.2μg/ml的终浓度加入细胞中,在37℃黑暗下培养15min。然后用pbs溶液清洗细胞1次,再加入培养基培养20至24小时。
[0039]
4.将细胞置于高通量共聚焦显微镜(moleculardevicesimagexpress)中,从每个孔捕获细胞的4个图像视野,对于每个样品,捕获3种激光波长图像:细胞的亮视野图像,显示总细胞核的hoechst染色图像和显示用质粒dna成功转染并表达gfp的gfp图像。用metaxpress软件对获得的hoechst染色图像和gfp图像分别进行细胞计数,再将表达gfp的细胞数除以总细胞核数,即为细胞绝对转染效率。绝对转染效率如下计算:。
[0040]
结果:部分化合物库对hela细胞的egfp
‑
mrna的转染效率示于表2中。
[0041]
表2:部分化合物库对hela细胞的egfp
‑
mrna的转染效率
实施例8:氨基脂质化合物制备的脂质纳米颗粒在bmdc原代细胞上的转染制剂方法:同实施例7。
[0042]
动物准备:选取6周龄的雌性c57bl/6小鼠,体重在20 g左右,饲养环境为spf级的饲养室,动物试验严格按照国家健康机构的指南以及动物伦理要求进行。
[0043]
细胞获取:把c57bl/6小鼠进行脱颈臼除死,并置于75%酒精中浸泡5分钟进行消毒,解刨获取小鼠大腿胫骨,并把附着的肌肉剔除露出骨质,然后用1ml吸有pbs的注射器把胫骨中的骨髓吹出,把所得骨髓吹散后通过50um滤网滤去杂质,往所得过滤物中加入红细胞裂解液后放置5分钟后进行100g、5分钟的离心除去上清液,将所得细胞置于1640培养基(含10%胎牛血清、20ng/ml gmcsf、10ng/ml il4)中重悬,并接种于6孔板中,接种密度为100000个细胞/毫升培养基,放置于37℃、5%co2细胞培养箱中,每2天进行半换液一次,于第七天收集悬浮细胞和松散贴壁的细胞,并接种到96孔全白酶标板接种密度为每孔10000个细胞,培养基体积为100ul。
[0044]
细胞转染:往铺有原代细胞的96孔全白酶标板中加入包裹萤光素酶mrna的脂质纳米颗粒,控制每孔中的萤光素酶mrna脂质纳米颗粒加入量为3ug。随后放置在37℃、5%co2浓度的培养箱中12小时,使萤光素酶mrna充分表达。
[0045]
转染效率检测:往96孔全白酶标板中每孔加入10ul的10mg/ml的d
‑
萤光素钾盐,马上放置于酶标仪中检测发光强度。代表性氨基脂质化合物在bmdc上转染fluc mrna的表达
强度见表3。dlin
‑
mc3作为对照,所述的氨基脂质多个与mc3表达强度相似,并有多个显著优于阳性对照。
[0046]
表3:代表性氨基脂质化合物在bmdc上的转染的表达强度
实施例9 胺基脂质化合物制备的脂质纳米颗粒的萤光素酶mrna体内递送性能评价1. 脂质纳米颗粒的制备制剂方法1:将本发明的胺基脂质化合物与dope、胆固醇、(1
‑ꢀ
(单甲氧基聚乙二醇)
ꢀ‑
2, 3 二肉豆寇酰基甘油(peg2000
‑
dmg)按45 : 10 : 42.5 : 2.5的摩尔比混合并溶解在无水乙醇中,使胺基脂质化合物的摩尔浓度为0.001
‑
0.01 mmol/l。使用微量注射泵,使所得的乙醇溶液和溶解有fluc
‑
mrna (trilink) 的醋酸钠溶液(50 mm, ph = 4.0)以1:3的体积比在微流道芯片中混合以制得脂质纳米颗粒的粗溶液,然后将其用透析盒(fisher,mwco 20,000)在1 x pbs、控温4 ℃下透析6h,在使用前通过0.22 μm的微孔滤膜过滤。胺基脂质化合物与萤光素酶mrna (fluc mrna)的质量比约为10 : 1。所得的脂质纳米颗粒(lnp)溶液通过皮下给药方式施用于受试动物。
[0047]
脂质纳米颗粒的表征:粒径的表征:所制备的脂质纳米颗粒的粒径和pdi通过nano
‑
zszen3600 (malvern)测定。取lnp溶液40 ul进行粒径测量,循环三次,每次循环30s。
[0048]
包封率检测:使用qubit
® rna hs assay试剂盒检测lnp rna浓度。理论rna浓度为投入的总rna量除以最终溶液的总体积。
[0049]
。
[0050]
表4:使用制剂方法1用代表性胺基脂质化合物制备的lnp的表征数据 氨基脂质编号z
‑
average(d.nm)pdi包封率1a4n4850.0698.7%2a3n10800.0299.0%3a12n4850.0399.0%4a12n91050.0298.5%5a14n13900.0697.2%6a15n221230.1099.6%7a14n161220.0499.0%8a26n161180.0398.3%9a21n16900.0298.8%10a32n231160.0599.3%11a34n22980.0499.2%12a35n211000.0399.1%13dlin
‑
mc31200.0898.4%制剂方法2:制备方法同制剂方法1,除了使用摩尔比为50 : 10 : 38.5 : 1.5的胺基脂质化合物、dspc、胆固醇和peg2000
‑
dmg。所得的脂质纳米颗粒(lnp)溶液通过尾静脉和肌肉注射的给药方式施用于受试动物。
[0051]
表5:使用制剂方法2用代表性胺基脂质化合物制备的lnp的表征数据 胺基脂质编号z
‑
average(d.nm)pdi包封率1a4n41270.0498.6%2a3n10950.0598.3%3a12n4960.0498.2%4a12n9920.0398.5%5a14n131200.0598.9%6a15n22950.0398.3%7a14n161300.0899.2%8a26n16900.0798.4%9a21n161150.0499.0%10a32n231250.0599.5%11a34n22880.0499.0%12a35n211100.0699.3%13dlin
‑
mc31200.0799.0%2. 动物实验
动物准备:选取体重约20 g的6周龄的雌性c57bl/6小鼠,于spf级的饲养室中饲养。动物试验严格按照国家健康机构的指南以及动物伦理要求进行。
[0052]
体内递送:每组随机选取9只c57bl/6小鼠,按0.5 mg/kg mrna的用量,分别使用皮下、肌肉和尾静脉注射三种给药方式注射脂质纳米颗粒溶液(每种给药方式3只小鼠)。12小时后,往每只小鼠体内通过尾静脉注射200 μl 10 mg/ml的d
‑
萤光素钾盐,10分钟后,将小鼠放置于活体成像系统(ivis
‑
200, xenogen)下,观察每只小鼠总的萤光强度,并拍照记录下来。代表性胺基脂质化合物通过3种给药方式递送的fluc mrna的表达强度见表6
‑
8。dlin
‑
mc3作为对照。
[0053]
表6:代表性胺基脂质化合物皮下给药递送的fluc mrna的表达强度 氨基脂质编号萤光强度1a4n42.7e+062a3n101.9e+073a12n47.6e+064a12n91.4e+075a14n133.7e+086a15n224.8e+087a14n163.1e+078a26n161.1e+079a21n168.2e+0610a32n239.7e+0611a34n221.9e+0712a35n216.8e+0613dlin
‑
mc33.1e+06表7:代表性胺基脂质化合物肌注给药递送的fluc mrna的表达强度 氨基脂质编号萤光强度1a4n44.1e+062a3n102.7e+063a12n41.2e+074a12n94.3e+075a14n132.8e+076a15n222.7e+077a14n164.7e+068a26n164.7e+069a21n168.2e+0610a32n234.2e+0611a34n229.1e+0612a35n211.8e+0613dlin
‑
mc38.5e+06表8:代表性胺基脂质化合物尾静脉给药递送的fluc mrna的表达强度
ꢀ
氨基脂质编号萤光强度1a4n44.6e+062a3n105.2e+063a12n43.6e+064a12n95.1e+075a14n137.2e+066a15n225.1e+077a14n165.2e+068a26n162.1e+069a21n166.5e+0610a32n233.9e+0611a34n222.1e+0612a35n218.2e+0613dlin
‑
mc32.7e+07实施例10:氨基脂质化合物制备的脂质纳米颗粒在bmdc原代细胞上免疫评价制剂方法:将本发明中所述的氨基脂质化合物与dope, 胆固醇, peg2000
‑
dmg的摩尔比为45:10:42.5:2.5的比例混合溶解在无水乙醇中。卵清蛋白mrna(ova mrna)溶解在醋酸钠溶液(50 mm, ph = 4.0)中。使用两个微量注射泵,控制其配比为乙醇溶液与醋酸钠溶液(50 mm, ph = 4.0)的比例为1:3,在微流道芯片中制得脂质纳米颗粒的粗溶液,再使用透析盒(fisher,mwco 20,000)在1x pbs、控温4 ℃下透析6h,使用前用0.22 μm的微孔滤膜过滤。氨基脂质化合物与卵清蛋白mrna(ova mrna)的质量比约为8:1。
[0054]
动物准备:选取6周龄的雌性c57bl/6小鼠,体重在20 g左右,饲养环境为spf级的饲养室,动物试验严格按照国家健康机构的指南以及动物伦理要求进行。
[0055]
细胞获取:同实施例8。
[0056]
免疫细胞的激活:往12孔板中每孔加入1ug卵清蛋白mrna脂质纳米颗粒,置于37℃,5%co2培养箱中培养24小时。用pbs溶液将细胞吹打下来,并用pbs洗涤离心(100g、5分钟)三次,之后用cd11c
‑
apc抗体和siinfekl
‑
h
‑
2kb
‑
pe抗体、cd11c
‑
apc抗体和mhc
‑
ii
‑
pe抗体进行孵育30分钟,然后用pbs洗涤离心(100g、5分钟)一次,除去没有结合上的抗体,然后用流式细胞仪(贝克曼cytoflex lx)检测。其中cd11是bmdc的标记物,因此cd11c
‑
apc抗体用于dc群体的标记,siinfekl
‑
h
‑
2kb
‑
pe抗体用于标记细胞群中呈递ova抗原的细胞群体,mhc
‑
ii
‑
pe抗体用于标记成熟dc细胞群体。结果如图1、2所示,a14n13和a15n12与mc3刺激bmdc成熟和呈递ova抗原能力相当,而a32n22,a34n22和a35n20免疫激活效果显著优于mc3对照组。
[0057]
实施例11:氨基脂质化合物制备的脂质纳米颗粒的萤光素酶mrna体内递送性能评价制剂方法:将本发明中所述的氨基脂质化合物与dope, 胆固醇, peg2000
‑
dmg的摩尔比为45:10 :42.5:2.5的比例混合溶解在无水乙醇中。萤光素酶mrna(fluc mrna)溶解在醋酸钠溶液(50 mm, ph = 4.0)中。使用两个微量注射泵,控制其配比为乙醇溶液与醋酸钠溶液(50 mm, ph = 4.0)的比例为1:3,在微流道芯片中制得脂质纳米颗粒的粗溶液,再
使用透析盒(fisher,mwco 20,000)在1x pbs、控温4 ℃下透析6h,使用前用0.22 μm的微孔滤膜过滤。氨基脂质化合物与萤光素酶mrna(fluc mrna)的质量比约为8:1。
[0058]
动物准备:选取6周龄的雌性c57bl/6小鼠,体重在20 g左右,饲养环境为spf级的饲养室,动物试验严格按照国家健康机构的指南以及动物伦理要求进行。
[0059]
体内递送:每组随机选取3只小鼠,按0.5 mg/kg的用量,使用皮下注射脂质纳米颗粒。6小时后,分别往每只小鼠体内通过尾静脉注射200 μl 10 mg/ml的d
‑
萤光素钾盐,10分钟后,将小鼠放置于活体成像系统(ivis
‑
200, xenogen)下,观察每只小鼠总的萤光强度,并拍照记录下来。代表性氨基脂质化合物三种给药方式递送fluc mrna的表达强度见表9
‑
10,dlin
‑
mc3作为对照。所述的多个氨基脂质与dlin
‑
mc3表达强度相似,并有多个显著优于阳性对照。
[0060]
表9:使用制剂方法2用代表性胺基脂质化合物制备的lnp的表征数据 胺基脂质编号z
‑
average(d.nm)pdi包封率1a4n4117.80.0598.9%2a3n10110.90.0698.6%3a12n4109.80.0498.6%4a12n9140.30.0498.9%5a14n13133.90.0698.5%6a15n22113.20.0598.8%7a14n16131.20.0799.2%8a26n16127.50.0898.5%9a21n16124.70.0699.0%10a32n23134.10.0499.3%11a34n22115.60.0899.2%12a35n21136.80.0599.0%13dlin
‑
mc3125.40.0898.6%表10:代表性氨基脂质化合物肌注给药递送fluc mrna的表达强度编号胺基脂质编号萤光强度1a4n42.8e+062a3n101.8e+063a12n42.1e+054a12n94.5e+065a14n135.1e+066a15n222.8e+067a14n168.1e+068a26n167.8e+069a21n166.1e+0510a32n234.1e+0611a34n221.2e+0712a35n216.4e+06
13dlin
‑
mc34.2e+06实施例12:氨基脂质化合物制备的脂质纳米颗粒的卵清蛋白mrna体内递送及免疫性能评价制剂方法:将本发明中所述的氨基脂质化合物与dope, 胆固醇, peg2000
‑
dmg的摩尔比为45:10:42.5:2.5的比例混合溶解在无水乙醇中。卵清蛋白mrna(ova mrna)溶解在醋酸钠溶液(50 mm, ph = 4.0)中。使用两个微量注射泵,控制其配比为乙醇溶液与醋酸钠溶液(50 mm, ph = 4.0)的比例为1:3,在微流道芯片中制得脂质纳米颗粒的粗溶液,再使用透析盒(fisher,mwco 20,000)在1x pbs、控温4 ℃下透析6h,使用前用0.22 μm的微孔滤膜过滤。氨基脂质化合物与卵清蛋白mrna(ova mrna)的质量比约为8:1。
[0061]
动物准备:选取6周龄的雌性c57bl/6小鼠,体重在20 g左右,饲养环境为spf级的饲养室,动物试验严格按照国家健康机构的指南以及动物伦理要求进行。
[0062]
体内递送:每组随机选取3只小鼠,按0.5 mg/kg的用量,使用皮下注射脂质纳米颗粒(day 0)。7天后,使用相同的量再加强一次(day 7)。在第21天尾静脉取血进行血清学分析,dlin
‑
mc3作为对照。
[0063]
酶联免疫吸附测定(elisa):对平底96孔板(nunc)进行预涂在50 mm碳酸盐缓冲液中,ova蛋白的浓度为每孔0.5
ꢀµ
g蛋白(ph 9.6)在4℃过夜,然后用5%甘氨酸封闭。使用pbs
‑
0.05%tween(pbs
‑
t,ph 7.4)将免疫动物的血清从10
‑2稀释至10
‑6,添加到孔中并在37℃下孵育1小时。辣根过氧化物酶(hrp)偶联的山羊抗小鼠igg在pbs
‑
t
‑
1%bsa中以1:10,000的稀释度进行标记。添加hrp基板后,在一个波长下确定光密度elisa酶标仪(bio
‑
rad)中检测450 nm下的吸光度。如图3所示,a14n13与mc3所产生的igg抗体滴定相当,而a32n22,a34n22,a35n20的igg抗体滴定显著优于mc3对照组。
[0064]
实施例13:氨基脂质化合物制备的脂质纳米颗粒的体内免疫和肿瘤治疗效果评价制剂方法:将本发明中所述的氨基脂质化合物与dope, 胆固醇, peg2000
‑
dmg的摩尔比为45:10:42.5:2.5的比例混合溶解在无水乙醇中。卵清蛋白mrna(ova mrna)溶解在醋酸钠溶液(50 mm, ph = 4.0)中。使用两个微量注射泵,控制其配比为乙醇溶液与醋酸钠溶液(50 mm, ph = 4.0)的比例为1:3,在微流道芯片中制得脂质纳米颗粒的粗溶液,再使用透析盒(fisher,mwco 20,000)在1x pbs、控温4 ℃下透析6h,使用前用0.22 μm的微孔滤膜过滤。氨基脂质化合物与卵清蛋白mrna(ova mrna)的质量比约为8:1。
[0065]
动物准备:选取6周龄的雌性c57bl/6小鼠,体重在20 g左右,饲养环境为spf级的饲养室,动物试验严格按照国家健康机构的指南以及动物伦理要求进行。
[0066]
体内递送:将 b16
‑
ova 黑色素瘤细胞 (1.5
ꢀ×ꢀ
10
5 ) 皮下注射到 4
‑
6 周龄的小鼠右侧。当肿瘤大小小于 50 mm3时(约在肿瘤接种后第 4 天或第 5 天)开始接种疫苗。通过肌肉注射含有 15
ꢀµ
g ova
‑
mrna 的lnp 制剂对动物进行免疫。使用数显卡尺每周测量肿瘤生长 3 次,计算公式为 0.5
ꢀ×ꢀ
长度
ꢀ×ꢀ
宽度。当肿瘤体积达到 2,000 mm3时对小鼠实施安乐死。将肿瘤抑制与携带新鲜接种肿瘤的小鼠进行比较。未注射疫苗组的中位生存期为29周,注射疫苗后,对应的中位生存期分别为:38周(mc3), 49周(a32n22), 52周(a35n20),如图4所示。
[0067]
以上所述仅为本发明专利的较佳实施例而已,并不用以限制本发明专利,凡在本发明专利的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明专利
的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1