一种含直链烯烃的二苯甲基哌嗪类化合物及其制备方法与流程
1.本发明实施例涉及医药技术领域,具体涉及一种含直链烯烃的二苯甲基哌嗪类化合物及其制备方法。
背景技术:
2.二苯甲基哌嗪类化合物可作为抗组胺类药物、阿片类镇疼药物,治疗或缓解皮炎、眩晕、中风、糖尿病以及中度疼痛等疾病(us20090176792a1);该类化合物还可作为关键中间体,合成调节钙通道活动药物、抗癌药物,针对性的治疗先天性偏头痛、心绞痛、癫痫、高血压、以及部分心律失常、肥胖等疾病(wo2007071035a1,wo2004032874a2,us2009124620a1,wo2013103973a1,acs chemical biology,2013,8(7):1590
‑
1599.),是一类应用广泛,潜力巨大的杂环类化合物。然而,随着该类化合物在医药领域应用的深入,药物效力的降低以及用药副作用越来越明显,不断开发新的二苯甲基哌嗪类化合物作为药物活性分子或活性分子砌块,来满足药物的高质量发展是常态。
3.基于直链烯烃化合物在现有药物中的活性(journal of medicinal chemistr y,2003,46:623
‑
633.),以及其自身的独特性质,苯环上引入直链烯烃,可提高药物分子的脂溶性,有望进一步促进药物分子在生物体内的药效释放,提高药物的生物活性,从而为相应药物的开发提供潜在的优质活性分子砌块。
4.由于现有技术中,含直链烯烃的新二苯甲基哌嗪类化合物的合成未见报道,对其合成路线进行研究是十分必要的。
技术实现要素:
5.为此,本发明实施例提供一种含直链烯烃的二苯甲基哌嗪类化合物及其制备方法,以解决上述技术问题。
6.为了实现上述目的,本发明实施例提供如下技术方案:
7.根据本发明实施例的第一方面,提供了一种含直链烯烃的二苯甲基哌嗪类化合物,为抗组胺类药物、阿片类镇疼药物的研发提供新的潜在活性分子或分子砌块,从而有望获得高活性、低副作用的药物应用于医药治疗当中。上述含直链烯烃的二苯甲基哌嗪类化合物的结构通式为以下式(1):
8.9.其中,
10.r1选自氢;
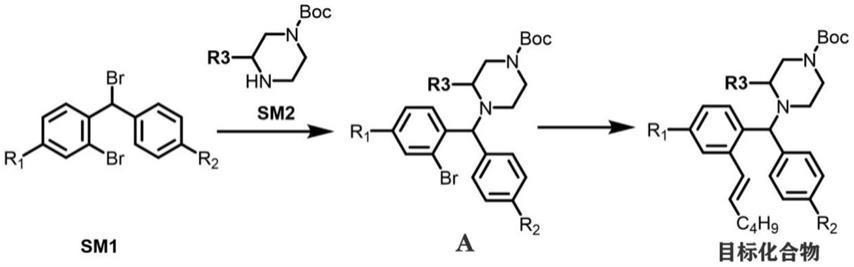
11.r2选自氢;
12.r3选自c2
‑
c6
‑
烯基、c1
‑
c6
‑
醛基、c1
‑
c6
‑
烷基氧酰基、c1
‑
c6
‑
烷基羰基、c1
‑
c6
‑
烷基氨基、c1
‑
c6
‑
烷基氨基羰基、c1
‑
c6
‑
烷基氨基磺酰基、c1
‑
c6
‑
烷基羟基、氨基氰基、二(氰基)
‑
c1
‑
c6
‑
烷基,包含至多4个相同或不同卤原子的c2
‑
c6
‑
卤代烯基、c1
‑
c6
‑
卤代烷基醛基、c1
‑
c6
‑
卤代烷基羰基中的一种。
13.进一步地,所述c1
‑
c6
‑
烷基氨基磺酰基被多个相同或不同的r4取代基取代;r4选自c1
‑
c6
‑
烷烃、c2
‑
c6
‑
烯烃、c2
‑
c6
‑
炔烃、c3
‑
c6
‑
环烷烃、包含至多4个相同或不同卤原子的c1
‑
c6
‑
卤代烷烃/c2
‑
c6
‑
卤代烯烃/c3
‑
c6
‑
卤代环烷烃中的至少一种。
14.优选地,所述r3选自
‑
coh、
‑
ch2oh、
‑
ch2ococh3或
‑
ch=ch2。
15.根据本发明实施例的第二方面,提供了一种含直链烯烃的二苯甲基哌嗪类化合物的制备方法,所述方法包括以下步骤:
16.步骤一:将化合物sm1溶于第一溶剂中,加入化合物sm2及碱,体系回流反应至反应完全,反应结束,待体系温度冷至室温后过滤,旋干、纯化得到化合物a;
17.步骤二:将所述化合物a溶于第二溶剂中,依次加入1
‑
烯基己基硼酸、碱和催化剂、配体,氮气保护下回流反应至反应完全,体系旋干,得到目标化合物;
18.所述目标化合物的结构通式为以下式(1):
[0019][0020]
所述化合物sm1的结构通式为以下式(2):
[0021][0022]
所述化合物sm2的结构通式为以下式(3):
[0023][0024]
所述化合物a的结构通式为以下式(4):
[0025][0026]
其中,
[0027]
r1选自氢;
[0028]
r2选自氢;
[0029]
r3选自c2
‑
c6
‑
烯基、c1
‑
c6
‑
醛基、c1
‑
c6
‑
烷基氧酰基、c1
‑
c6
‑
烷基羰基、c1
‑
c6
‑
烷基氨基、c1
‑
c6
‑
烷基氨基羰基、c1
‑
c6
‑
烷基氨基磺酰基、c1
‑
c6
‑
烷基羟基、氨基氰基、二(氰基)
‑
c1
‑
c6
‑
烷基,包含至多4个相同或不同卤原子的c2
‑
c6
‑
卤代烯基、c1
‑
c6
‑
卤代烷基醛基、c1
‑
c6
‑
卤代烷基羰基中的一种。
[0030]
进一步地,所述c1
‑
c6
‑
烷基氨基磺酰基被多个相同或不同的r4取代基取代;r4选自c1
‑
c6
‑
烷烃、c2
‑
c6
‑
烯烃、c2
‑
c6
‑
炔烃、c3
‑
c6
‑
环烷烃、包含至多4个相同或不同卤原子的c1
‑
c6
‑
卤代烷烃/c2
‑
c6
‑
卤代烯烃/c3
‑
c6
‑
卤代环烷烃中的至少一种。
[0031]
进一步地,在所述步骤一中,所述第一溶剂选自乙腈、二氯乙烷、四氢呋喃、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、乙酸钾、乙酸钠、n,n
‑
二异丙基乙胺中的至少一种;所述化合物sm1与所述化合物sm2、所述碱的摩尔比为1:(0.8
‑
1.2):(1.5
‑
3.5);所述反应时间为4
‑
8h。
[0032]
进一步地,在所述步骤二中,所述第二溶剂选自甲苯、二甲基亚砜、n,n
‑
二甲基甲酰胺、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸铯、碳酸钠中的至少一种;所述催化剂选自pd2(dba)3、pd(oac)2、nicl2(pcy3)2;所述配体选自三环己基膦或1,3
‑
双(二环己膦基)丙烷;所述化合物a与1
‑
烯基己基硼酸、碱、催化剂、配体的摩尔比为1:(1.1
‑
1.5):(3
‑
5):(0.05
‑
0.15):(0.05
‑
0.15);反应时间为8
‑
15h。
[0033]
根据本发明实施例的第三方面,提供了一种含直链烯烃的二苯甲基哌嗪类化合物的制备方法,所述方法包括以下步骤:
[0034]
步骤一:化合物sm1溶于第一溶剂中,加入化合物sm2及碱,体系回流反应至反应完全,反应结束,待体系温度冷至室温后过滤,旋干、纯化得到化合物b1;
[0035]
步骤二:将所述化合物b1溶于第二溶剂中,依次加入1
‑
烯基己基硼酸、碱和催化剂、配体,氮气保护下回流反应至反应完全,体系旋干,得到化合物b2;
[0036]
步骤三:将所述化合物b2溶于有第三溶剂中,冰水浴下加入碱,加毕升至室温反应2
‑
4h,过滤后旋干溶剂,乙酸乙酯溶解,饱和食盐水洗,无水硫酸钠干燥,过滤,滤液旋干得到化合物b3;
[0037]
步骤四:将所述化合物b3溶于第四溶剂中,冰水浴下加入氧化剂,加毕后升至室温反应1
‑
2h,淬灭反应,水层用二氯甲烷萃取,合并有机层,饱和食盐水洗涤,干燥,过滤,滤液浓缩得到化合物b4;
[0038]
步骤五:将甲基三苯基溴化膦溶于第五溶剂中,
‑
5~0℃下滴入碱,滴毕后保温搅拌,而后加入所述化合物b4溶于所述第五溶剂的溶液,保温反应至反应完全,用饱和氯化铵淬灭反应,水层用乙酸乙酯萃取,合并有机层用饱和食盐水洗,无水硫酸钠干燥,过滤,滤液浓缩,硅胶柱层析得到目标化合物;
[0039]
所述目标化合物的结构通式为以下式(1):
[0040][0041]
所述化合物sm1的结构通式为以下式(2):
[0042][0043]
所述化合物sm2的结构通式为以下式(5):
[0044][0045]
所述化合物b1的结构通式为以下式(6):
[0046][0047]
所述化合物b2的结构通式为以下式(7):
[0048][0049]
所述化合物b3的结构通式为以下式(8):
[0050][0051]
所述化合物b4的结构通式为以下式(9):
[0052][0053]
其中,
[0054]
r1选自氢;
[0055]
r2选自氢;
[0056]
r3选自
‑
ch=ch2。
[0057]
进一步地,在所述步骤一中,所述第一溶剂选自乙腈、二氯乙烷、四氢呋喃、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、乙酸钾、乙酸钠、n,n
‑
二异丙基乙胺中的至少一种;所述化合物sm1与所述化合物sm2、所述碱的摩尔比为1:(0.8
‑
1.2):(1.5
‑
3.5);所述反应时间为4
‑
8h。
[0058]
进一步地,在所述步骤二中,所述第二溶剂选自甲苯、二甲基亚砜、n,n
‑
二甲基甲酰胺、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸铯、碳酸钠中的至少一种;所述催化剂选自pd2(dba)3、pd(oac)2、nicl2(pcy3)2;所述配体选自三环己基膦或1,3
‑
双(二环己膦基)丙烷;所述化合物b1与1
‑
烯基己基硼酸、碱、催化剂、配体的摩尔比为1:(1.1
‑
1.5):(3
‑
5):(0.05
‑
0.15):(0.05
‑
0.15);所述反应时间为8
‑
15h。
[0059]
进一步地,在所述步骤三中,所述第三溶剂选自甲醇、乙醇、异丙醇、四氢呋喃、1,
4
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、氢氧化钠、氢氧化钾中的至少一种;所述化合物b2与碱的摩尔比为1:(2.5
‑
4);所述反应时间为2
‑
4h。
[0060]
进一步地,在所述步骤四中,所述第四溶剂选自二氯甲烷、二氯乙烷、四氢呋喃、氯仿中的至少一种;所述氧化剂选自戴斯
‑
马丁氧化剂、重铬酸吡啶嗡盐、2
‑
碘酰基苯甲酸中的至少一种;所述化合物b3与氧化剂的摩尔比为1:(1.5
‑
3);所述反应时间为1
‑
2h。
[0061]
进一步地,在所述步骤五中,所述第五溶剂选自四氢呋喃、n,n
‑
二甲基甲酰胺、二甲基亚砜中的至少一种;所述碱选自正丁基锂、乙醇钠、叔丁醇钠、二甲基亚砜盐中的至少一种;化合物b4与甲基三苯基溴化膦、碱的摩尔比为1:(1.5
‑
2.5):(1.5
‑
2.5);加入碱后,保温搅拌10
‑
50min;所述化合物b4溶于所述第五溶剂的溶液,保温反应0.5
‑
1.5h。
[0062]
本发明实施例具有如下优点:
[0063]
本发明实施例提供了一种含直链烯烃的二苯甲基哌嗪类化合物及其制备方法,期望直接作用于病灶或作为分子砌块参与到新药研发当中。所述含直链烯烃的新二苯甲基哌嗪类化合物的制备方法,以1
‑
溴
‑2‑
(溴(苯基)甲基)苯为原料,历经亲核取代、碳碳偶联两步反应得到目标化合物。整条制备路线反应稳固,所用条件温和,操作简便,并且碳碳偶联反应通过优选催化体系,显著提高了反应活性,反应收率高,可为该类化合物的合成提供可靠参考。
具体实施方式
[0064]
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。除非另作定义,此处使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。
[0065]
下面通过具体实施例对本发明进行详细描述,但不限于此。
[0066]
本发明实施例公开了一种含直链烯烃的二苯甲基哌嗪类化合物,该含直链烯烃的二苯甲基哌嗪类化合物的结构通式为以下式(1):
[0067][0068]
其中,
[0069]
r1选自氢;
[0070]
r2选自氢;
[0071]
r3选自c2
‑
c6
‑
烯基、c1
‑
c6
‑
醛基、c1
‑
c6
‑
烷基氧酰基、c1
‑
c6
‑
烷基羰基、c1
‑
c6
‑
烷
基氨基、c1
‑
c6
‑
烷基氨基羰基、c1
‑
c6
‑
烷基氨基磺酰基、c1
‑
c6
‑
烷基羟基、氨基氰基、二(氰基)
‑
c1
‑
c6
‑
烷基,包含至多4个相同或不同卤原子的c2
‑
c6
‑
卤代烯基、c1
‑
c6
‑
卤代烷基醛基、c1
‑
c6
‑
卤代烷基羰基中的一种。
[0072]
其中,c1
‑
c6
‑
烷基氨基磺酰基可以被多个相同或不同的r4取代基取代;r4选自c1
‑
c6
‑
烷烃、c2
‑
c6
‑
烯烃、c2
‑
c6
‑
炔烃、c3
‑
c6
‑
环烷烃、包含至多4个相同或不同卤原子的c1
‑
c6
‑
卤代烷烃/c2
‑
c6
‑
卤代烯烃/c3
‑
c6
‑
卤代环烷烃中的至少一种。
[0073]
由于二苯甲基哌嗪类药物的使用越来越深入,药效降低、副作用明显等问题越来越突出,二苯甲基哌嗪类化合物活性分子谱需不断更新是药物发展的常态。本发明提出的二苯甲基哌嗪类新化合物可作为针对多种疾病的潜在活性分子,由于二苯甲基哌嗪类化合物的哌嗪环部分在许多疾病治疗中,作为活性基团起作用,结合支链烯烃在现代医药当中的生物活性,运用药物设计策略之一的片段拼接,将直链烯烃引入苯环部分中,有望提高药物在生物体内的释放程度,并进一步的提高药物的药效;同时,为获得生物利用度高的生物活性分子,在哌嗪环上引入多种不同基团,通过协同作用,进行水溶性与脂溶性平衡度的取舍,从而获得高潜力的活性药物活性中间体。
[0074]
本发明实施例还公开了上述一种含直链烯烃的二苯甲基哌嗪类化合物的制备方法。具有式(1)作为结构通式的上述二苯甲基哌嗪类化合物的合成,采用以下合成路线:
[0075][0076]
上述一种含直链烯烃的二苯甲基哌嗪类化合物的制备方法,具体地包括以下步骤:
[0077]
步骤一:将化合物sm1溶于第一溶剂中,加入化合物sm2及碱,体系回流反应至反应完全,反应结束,待体系温度冷至室温后过滤,旋干、纯化得到化合物a;
[0078]
步骤二:将所述化合物a溶于第二溶剂中,依次加入1
‑
烯基己基硼酸、碱和催化剂、配体,氮气保护下回流反应至反应完全,体系旋干,得到目标化合物。
[0079]
在上述步骤一中,所述第一溶剂选自乙腈、二氯乙烷、四氢呋喃、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、乙酸钾、乙酸钠、n,n
‑
二异丙基乙胺中的至少一种;所述化合物sm1与所述化合物sm2、所述碱的摩尔比为1:(0.8
‑
1.2):(1.5
‑
3.5)所述反应时间为4
‑
8h。
[0080]
在上述步骤二中,所述第二溶剂选自甲苯、二甲基亚砜、n,n
‑
二甲基甲酰胺、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸铯、碳酸钠中的至少一种;所述催化剂选自pd2(dba)3、pd(oac)2、nicl2(pcy3)2;所述配体选自三环己基膦或1,3
‑
双(二环己膦基)丙烷;所述反应时间为8
‑
15h。进一步地,化合物a与1
‑
烯基己基硼酸、碱、催化剂、配体的摩尔比为1:(1.1
‑
1.5):(3
‑
5):(0.05
‑
0.15):(0.05
‑
0.15)。
[0081]
另外,在本发明实施例中,当上述r3选自c2
‑
c6稀基、c1
‑
c6
‑
醛基、c1
‑
c6
‑
烷基氧酰
基、c1
‑
c6烷基羟基;上述r3选自
‑
coh、
‑
ch2oh、
‑
ch2ococh3或
‑
ch=ch2。
[0082]
以下,以r1选自氢;r2选自氢;r3选自
‑
ch=ch2为例,说明本发明实施例中的上述一种含直链烯烃的二苯甲基哌嗪类化合物的制备方法,此时可采用以下合成路线:
[0083][0084]
其具体包括以下步骤:
[0085]
步骤一:化合物sm1溶于第一溶剂中,加入化合物sm2及碱,体系回流反应至反应完全,反应结束,待体系温度冷至室温后过滤,旋干、纯化得到化合物b1;
[0086]
步骤二:将所述化合物b1溶于第二溶剂中,依次加入1
‑
烯基己基硼酸、碱和催化剂、配体,氮气保护下回流反应至反应完全,体系旋干,得到化合物b2;
[0087]
步骤三:将所述化合物b2溶于有第三溶剂中,冰水浴下加入碱,加毕升至室温反应2
‑
4h,过滤后旋干溶剂,乙酸乙酯溶解,饱和食盐水洗,无水硫酸钠干燥,过滤,滤液旋干得到化合物b3;
[0088]
步骤四:将所述化合物b3溶于第四溶剂中,冰水浴下加入氧化剂,加毕后升至室温反应1
‑
2h,淬灭反应,水层用二氯甲烷萃取,合并有机层,饱和食盐水洗涤,干燥,过滤,滤液浓缩得到化合物b4;
[0089]
步骤五:将甲基三苯基溴化膦溶于第五溶剂中,
‑
5~0℃下滴入碱,滴毕后保温搅拌,而后加入所述化合物b4溶于所述第五溶剂的溶液,保温反应至反应完全,用饱和氯化铵淬灭反应,水层用乙酸乙酯萃取,合并有机层用饱和食盐水洗,无水硫酸钠干燥,过滤,滤液浓缩,硅胶柱层析得到目标化合物。
[0090]
由于现有技术中,缺乏含直链二苯甲基哌嗪的合成的方法的相关报道,本发明就该化合物的合成提供了一种可靠的合成方法。以化合物sm1 1
‑
溴
‑2‑
(溴(苯基)甲基)苯与带有对应基团的化合物sm2叔丁基哌嗪
‑1‑
羧酸酯衍生物作为原料进行亲核取代,一步构建二苯甲基哌嗪框架,从而降低二苯甲基哌嗪框架中哌嗪环上添加修饰基团的难度;而后在该框架上,进行直链烯烃化,由于现有技术中苯类衍生物的烯烃化,多通过格式试剂实现,而由于格氏试剂自身的特性,反应中需严格除水除氧,反应过程操作繁琐的同时,规模化扩生产存在诸多不便,这将在一定程度上限制该类化合物的应用;本发明通过对催化剂及配体的择优选择,采用(e)
‑1‑
己烯基二羟基硼烷处理所得二苯甲基哌嗪衍生物,历经碳碳偶联反应,添加直链烯烃获得目标化合物,反应中所用催化剂与优选的高活性配体进行金属转移,催化反应,降低了反应所需活化能,提高了反应活性,使反应以高收率实现,并且该反
应对基团的容忍度高,反应过程中无需严格除水除氧,反应便于控制,操作更具简便性,适用于多种直链二苯甲基哌嗪类化合物的合成。
[0091]
在上述步骤一中,第一溶剂选自乙腈、二氯乙烷、四氢呋喃、1,2
‑
二氧六环中的至少一种;碱选自碳酸钾、碳酸钠、乙酸钾、乙酸钠、n,n
‑
二异丙基乙胺中的至少一种;化合物sm1与化合物sm2、碱的摩尔比为1:(0.8
‑
1.2):(1.5
‑
3.5);反应时间为4
‑
8h。
[0092]
在上述步骤二中,第二溶剂选自甲苯、二甲基亚砜、n,n
‑
二甲基甲酰胺、1,2
‑
二氧六环中的至少一种;碱选自碳酸钾、碳酸铯、碳酸钠中的至少一种;催化剂选自pd2(dba)3、pd(oac)2、nicl2(pcy3)2;配体选自三环己基膦或1,3
‑
双(二环己膦基)丙烷,化合物b1与1
‑
烯基己基硼酸、碱、催化剂、配体的摩尔比为1:(1.1
‑
1.5):(3
‑
5):(0.05
‑
0.15):(0.05
‑
0.15),反应时间为8
‑
15h。
[0093]
在上述步骤三中,第三溶剂选自甲醇、乙醇、异丙醇、四氢呋喃、1,4
‑
二氧六环中的至少一种;碱选自碳酸钾、碳酸钠、氢氧化钠、氢氧化钾中的至少一种;化合物b2与碱的摩尔比为1:(2.5
‑
4);反应时间为2
‑
4h,进一步优选2h。
[0094]
在上述步骤四中,第四溶剂选自二氯甲烷、二氯乙烷、四氢呋喃、氯仿中的至少一种;氧化剂选自戴斯
‑
马丁氧化剂(dess
‑
martin)、重铬酸吡啶嗡盐(pyridinium dichromate)、2
‑
碘酰基苯甲酸中的至少一种;并进一步优选dess
‑
martin作为氧化试剂;化合物b3与氧化剂的摩尔比为1:(1.5
‑
3);反应时间为1
‑
2h,并进一步优选为1h。
[0095]
在上述步骤五中,第五溶剂选自四氢呋喃、n,n
‑
二甲基甲酰胺、二甲基亚砜中的至少一种;碱选自正丁基锂、乙醇钠、叔丁醇钠、二甲基亚砜盐中的至少一种;化合物b4与甲基三苯基溴化膦、碱的摩尔比为1:(1.5
‑
2.5):(1.5
‑
2.5);加入碱后,保温搅拌10
‑
50min;优选20min;所述化合物b4溶于所述第五溶剂的溶液滴毕后,保温反应0.5
‑
1.5h,优选0.5h。
[0096]
上述合成路线中,以化合物sm1 1
‑
溴
‑2‑
(溴(苯基)甲基)苯与3
‑
(乙酰氧基甲基)
‑4‑
((2
‑
溴苯基)(苯基)甲基)哌嗪
‑1‑
羧酸叔丁酯作为原料进行亲核取代,直接构建二苯甲基哌嗪框架,而后通过优选催化剂以及配体,降低反应进行所需活化能,使反应活性大大提高,实现直链烯烃基团的添加,该步反应基团容忍度高,可用于多种底物的合成,反应过程操作简便,无需严格除水除氧,利于规模化扩大生产;而后所得中间体碱性条件下实现水解;采用氧化试剂进行醇基的氧化,实现醇基到醛基的基团转化,得到含甲醛基的二苯甲基哌嗪化合物,该步优选dess
‑
martin作为氧化试剂,该试剂具有反应时间短、反应条件温和、氧化试剂用量少等特点,从而使提高了反应活性,缩短了反应时间,减少了反应所需的能耗以及生产的物料成本;最后在甲基三苯基溴化膦与碱作用下烯烃化,得到含乙烯基的二苯甲基哌嗪化合物,该反应副产物少,反应具有专一性;整条合成路线,化合物得到连续合成,有利于进一步降低生产成本以及提高工业生产的可行性,从而获得具有最高性价比的生产工艺路线。
[0097]
实施例1
[0098]
以下,以r1选自氢;r2选自氢;r3选自
‑
ch2oac;为例,说明本发明实施例中的上述一种含直链烯烃的二苯甲基哌嗪类化合物的制备方法。
[0099]
步骤一:化合物b1的合成
[0100][0101]
将化合物sm1(20g,61.34mmol,1eq)溶于乙腈(150ml)中,加入化合物sm2(15.85g,61.36mmol,1eq)及碳酸钾(17g,123.0mmol,2eq),体系回流反应4小时。反应结束,待体系温度冷至室温后过滤,旋干溶剂后硅胶柱层析得到白色固体27.51g,即化合物b1,收率87.4%,纯度98.1%。
[0102]
化合物b1的结构表征数据如下:
[0103]
[m+h]
+
=503.16
[0104]1h
‑
nmr(300mhz,cdcl3)δ7.79(m,1h),7.48(m,3h),7.28(m,3h),7.20(m,1h),7.06(m,1h),5.42(m,1h),4.28(m,2h),3.95(m,2h),3.08(m,3h),2.61(m,2h),2.00(m,3h),1.43(s,9h)。
[0105]
步骤二:化合物b2的合成
[0106][0107]
将化合物b1(27.51g,53.63mmol,1eq)溶于甲苯(300ml),依次加入1
‑
烯基己基硼酸(8.24g,64.39mmol,1.2eq)、碳酸钾(22.24g,160.91mmol,3eq)、pd2(dba)3(4.91g,5.365mmol,0.1eq)和1,3
‑
双(二环己膦基)丙烷(2.34g,5.365mmol,0.1eq),体系氮气置换三次。氮气保护下回流反应过夜。次日,体系旋干溶剂后硅胶柱层析得到淡黄色液体26.09g,即化合物b2,收率94.00%,纯度97.9%。
[0108]
化合物b2的结构表征数据如下:
[0109]
[m+h]
+
=507.32
[0110]1h
‑
nmr(300mhz,cdcl3)δ7.73(q,1h),7.38(d,j=5.7hz,2h),7.13
‑
7.35(m,6h),6.80(m,1h),5.91(m,1h),5.20(m,1h),4.30(m,2h),3.95(m,1h),3.0(m,2h),2.57(m,2h),2.22(q,2h),1.97(m,3h),1.49(q,2h),1.43(s,9h),1.38(m,4),0.98(dd,j=7.8hz,3h)。
[0111]
步骤三至步骤五:目标化合物的合成
[0112][0113]
将步骤二所得化合物b2(4.78g,9.43mmol,1eq)溶于甲醇(50ml)中,冰水浴下加入
碳酸钾(3.91g,28.29mmol,3eq),加毕后撤去冰水浴室温反应2小时。过滤后旋干溶剂,60ml乙酸乙酯溶解,饱和食盐水洗,无水硫酸钠干燥。过滤,减压蒸去溶剂后得到4.45g淡黄色液体,即化合物b3,收率为93.34%,纯度为92.00%。
[0114]
将上步所得淡黄色液体化合物b3(4.09g,8.10mmol)溶于二氯甲烷(40ml),冰水浴下分三次加dess
‑
martin氧化剂(8.6g,24.90mmol,2.5eq),加毕后撤去冰水浴室温反应1h。加入饱和碳酸氢钠和硫代硫酸钠的混合溶液淬灭反应后,水层用二氯甲烷萃取,合并有机层后用饱和食盐水洗,无水硫酸钠干燥。过滤,减压蒸去溶剂后,得到3.68g淡黄色液体,即化合物b4,收率为89.78%,纯度为91.30%。
[0115]
将甲基三苯磷溴(5.19g,14.54mmol,2eq)溶于四氢呋喃(120ml),0℃下慢慢滴入正丁基锂(5.80ml,14.54mmol,2eq),滴毕后0℃搅拌20min,后慢慢滴入上述得到的化合物b4(3.68g,7.27mmol,1eq)的四氢呋喃溶液(20ml),保温反应0.5h后,用饱和氯化铵淬灭反应。水层用乙酸乙酯萃取,合并有机层后用饱和食盐水洗,无水硫酸钠干燥。过滤,减压蒸去溶剂后,硅胶柱层析得到2.61g淡黄色液体,即目标化合物,收率为76.37%,纯度为98.12%。
[0116]
目标化合物的结构表征数据如下:
[0117]
[m+h]
+
=461.32
[0118]1h
‑
nmr(300mhz,cdcl3)δ7.63(m,1h),7.12
‑
7.34(m,9h),6.77(t,j=30.3,1h),6.39(m,2h),5.58(m,2h),4.88(s,1h),3.80(m,2h),3.32(m,3h),3.02(brs,1h),2.75(m,2h),1.40(s,9h),1.38(m,4),0.98(m,3h)。
[0119]
实施例2
‑
11及对比例1
‑5[0120]
实施例2
‑
11、对比例1
‑
5的步骤与上述实施例1相同,仅改变了步骤二的部分工艺条件。为使结果更加直观明了,实施例2
‑
11、对比例1
‑
5采用了列表的方式,对本发明进行了具体说明。
[0121]
表1反应条件对中间体化合物b2收率的影响
[0122][0123]
由表1,反应在高沸点溶剂中可以获得较为理想的反应收率,如上述实施例1
‑
4所示,优选溶剂为甲苯、二甲基亚砜、n,n
‑
二甲基甲酰胺、1,2
‑
二氧六环,当采用如对比例4所示的低沸点溶剂进行反应,则反应收率降低明显。由于反应中1
‑
烯基己基硼酸存在部分脱硼作用,其用量大于理论值的1eq时,反应收率较为理想,如实施例5
‑
6所示,其用量优选1.1
‑
1.5eq,当其用量降低至对比例5中的0.7eq时,原料剩余较多,收率显著降低;该步反应的实质为碳碳偶合,采用对比例1
‑
3所述催化剂pd(ph3p)4、pd(dppf)cl2、pd cl2(ph3p)2对反
应进行催化,所得反应结果并不理想,当催化体系更换至实施例1及实施例7
‑
9所示的pd2(dba)3、pd(oac)2与配体1,3
‑
双(二环己膦基)丙烷、三环己基膦的任一组合时,反应收率明显提高,并优选pd2(dba)3与1,3
‑
双(二环己膦基)丙烷催化体系;特别的,实施例10采用镍催化剂nicl2(pcy3)2作为催化剂,三环己基膦作为配体时,反应结果虽稍逊于pd2(dba)3与1,3
‑
双(二环己膦基)丙烷的催化体系,但反应结果同样优异,这为镍催化剂催化类似反应物的碳碳偶联提供了参考。该反应还可在多种不同碱中进行,如实施例1以及实施例11,采用碳酸钾或碳酸钠作为碱,较为优异的实现了本发明。
[0124]
由以上技术方案可知,本发明提出了一些含直链烯烃的二苯甲基哌嗪新化合物,可为二苯甲基哌嗪类药物的发展提供潜在的活性分子或活性分子砌块,从而进一步拓展该类活性药物的候选分子谱。本发明提出的含直链烯烃的二苯甲基哌嗪化合物的合成制备方法,可为该类化合物的合成提供可靠的参考,并且优选反应策略,采用带有相应基团的化合物sm2叔丁基哌嗪
‑1‑
羧酸酯衍生物作为原料构建二苯甲基哌嗪框架,降低了目标化合物的合成难度,直链烯烃的添加步骤,优选催化剂及配体进行反应,降低反应所需活化能,使反应活性提高、副反应减少,适用范围广,反应条件温和,操作简便,尤其是该步反应中,采用价格更为低廉的镍催化剂与相应配体催化反应,为镍催化剂催化类似反应物的碳碳偶联提供了参考;整个合成过程反应收率高,未用到高毒、高危试剂,反应后处理简单,合成步骤简短,具有绿色化学的特征。
[0125]
虽然,上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1