一种苯并吡嗪类化合物及其合成方法与流程
1.本发明实施例涉及医药技术领域,具体涉及具有多种药物活性的一种苯并吡嗪类化合物及其合成方法。
背景技术:
2.苯并杂环类化合物,可作为高活性人体害虫、动物寄生虫防治药物用于防治血吸虫、绦虫、囊虫、华支睾吸虫、肺吸虫、姜片虫等(如cn102267998a所述)。该类化合物还可作优异的杀菌剂,防治革兰氏阳性菌及革兰氏阴性菌等病菌(如cn101497612a所述)。此外,该类化合物还是选择性多巴胺能d1激动剂和拮抗剂,用于治疗震颤麻痹、强直或运动障碍等疾病(journal of heterocyclic chemistry,1991,28(5):1219
‑
1224.)。对于缓解松弛前列腺平滑肌的张力,从而缓解膀胱下梗阻症状,该类化合物也具有较为理想的活性(如gb2295387所述)。然而随着药物的长期使用,苯并杂环类化合物也无法避免抗性提高、副作用凸显这一药物发展的必然问题,这要求本领域研发人员不断开发新的活性分子,不断更新候选分子谱,以获得新型高效药物应对这一药物发展现状。
3.关于苯并杂环类化合物的合成,现有技术中报道较少,专利cn101497612a以2
‑
氟苯乙酸作为原料,历经合环、拓环、醚化、硝化、硝基还原、合哌嗪环以及脱甲酰乙酯基合成10
‑
氟
‑
1,2,3,4,4a,5,6,11
‑
八氢吡嗪并[1,2
‑
b][2]苯并氮杂,构建七元环的关键步骤在该路线中反应收率较低,仅为39.4%,且反应中使用了叠氮钠,该试剂属剧毒类化合物,反应中也易与大多数金属的盐类、氢氧化物或酸类生成极具爆炸性的有毒叠氮化合物,安全隐患高,不仅不适合工业化扩大生产,对于实验室的少量成品合成也存在诸多隐患。故而,研究一条操作简便、活性较高、所用试剂安全环保的苯并杂环类化合物的合成路线是十分必要的。
技术实现要素:
[0004]
为此,本发明实施例提供一种苯并吡嗪类化合物及其合成方法,以解决上述技术问题。
[0005]
为了实现上述目的,本发明实施例提供如下技术方案:
[0006]
根据本发明实施例的第一方面,提供了一种苯并吡嗪类化合物,具体为一种苯环取代的1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类化合物,从而拓展苯并吡嗪类化合物杀虫剂、杀菌剂的候选分子谱。上述苯并吡嗪类化合物的结构通式为以下式(1):
[0007][0008]
其中,
[0009]
r1选自氢、羟基、卤素、c1
‑
c3烷基、c1
‑
c2烷基氧羰基c1
‑
c2烷基中的一种;
[0010]
r2选自氢、羟基、卤素、c1
‑
c3烷基、c1
‑
c2烷基氧羰基c1
‑
c2烷基中的一种;
[0011]
r3选自环己基甲酰基、环戊甲酰基、环庚烷甲酰基、1
‑
环己烯
‑1‑
碳酰基、3
‑
环己烯
‑1‑
碳酰基、四氢吡喃
‑4‑
甲酰基中的一种。
[0012]
进一步地,所述r1选自氢、羟基、甲基、乙基、丙基、乙酸甲酯基中的一种;所述r2选自氢、羟基、甲基、乙基、丙基、乙酸甲酯基中的一种;所述r3选自环己基甲酰基、环戊甲酰基中的一种。
[0013]
根据本发明实施例的第二方面,提供了一种苯并吡嗪类化合物的合成方法,所述方法包括以下步骤:
[0014]
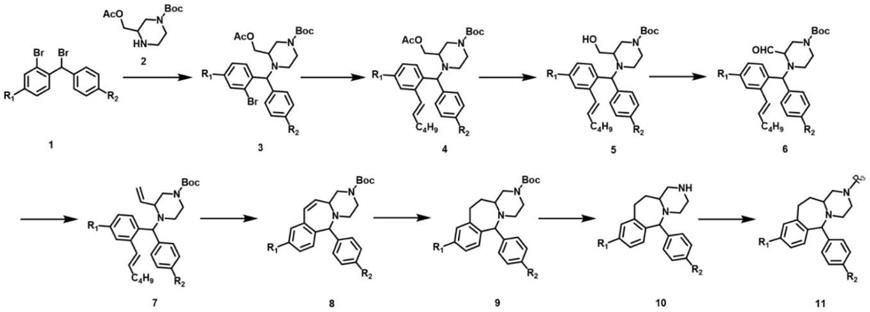
步骤一:将化合物1溶于第一溶剂中,加入化合物2及碱,体系回流反应至反应完全,反应结束,待体系温度冷至室温后过滤,旋干、纯化得到化合物3;
[0015]
步骤二:将所述化合物3溶于第二溶剂中,依次加入1
‑
烯基己基硼酸、碱和催化剂、配体,氮气保护下回流反应至反应完全,体系旋干、得到化合物4;
[0016]
步骤三:将所述化合物4溶于第三溶剂中,冰水浴下加入碱,加毕升至室温反应2
‑
4h,过滤后旋干溶剂,乙酸乙酯溶解,饱和食盐水洗,无水硫酸钠干燥,过滤,滤液旋干得到化合物5;
[0017]
步骤四:将所述化合物5溶于第四溶剂中,冰水浴下加入氧化剂,加毕后升至室温反应1
‑
2h;淬灭反应,水层用二氯甲烷萃取,合并有机层,饱和食盐水洗涤,干燥,过滤,滤液浓缩得到化合物6;
[0018]
步骤五:将甲基三苯基溴化膦溶于第五溶剂中,
‑
5~0℃下滴入碱,滴毕后保温搅拌,而后加入所述化合物6溶于所述第五溶剂的溶液,保温反应至反应完全,用饱和氯化铵淬灭反应,水层用乙酸乙酯萃取,合并有机层用饱和食盐水洗,无水硫酸钠干燥,过滤,滤液浓缩,硅胶柱层析得到化合物7;
[0019]
步骤六:将所述化合物7溶于第六溶剂中,加入催化剂,回流反应过夜至反应完全,减压旋干溶剂,硅胶柱纯化得到化合物8;
[0020]
步骤七:将所述化合物8溶于第七溶剂中,加入催化剂,室温氢化反应至完全;过滤,甲醇洗滤渣,滤液旋干溶剂后,硅胶柱纯化得到化合物9;
[0021]
步骤八:将所述化合物9溶于第八溶剂中,冰水浴下加入酸液,滴毕后撤去冰浴,室温反应至反应完全;慢慢加入饱和碳酸氢钠溶液淬灭反应后,二氯甲烷萃取后饱和食盐水洗,无水硫酸钠干燥;过滤,减压蒸去溶剂后得到化合物10,未经纯化,直接用于下步;
[0022]
步骤九:将上步所得化合物10溶于第九溶剂中,冰水浴下依次加入碱和甲酰氯衍生物,室温反应过夜至完全,后加入饱和碳酸氢钠溶液淬灭反应;水层用二氯甲烷萃取,合并有机层,无水硫酸钠干燥;旋干溶剂后,硅胶柱纯化得到目标化合物11;
[0023]
所述目标化合物的结构通式为以下式(11):
[0024][0025]
所述化合物1的结构通式为以下式(1):
[0026][0027]
所述化合物2的结构通式为以下式(2):
[0028][0029]
所述化合物3的结构通式为以下式(3):
[0030][0031]
所述化合物4的结构通式为以下式(4):
[0032]
[0033]
所述化合物5的结构通式为以下式(5):
[0034][0035]
所述化合物6的结构通式为以下式(6):
[0036][0037]
所述化合物7的结构通式为以下式(7):
[0038][0039]
所述化合物8的结构通式为以下式(8):
[0040][0041]
所述化合物9的结构通式为以下式(9):
[0042][0043]
[0044]
所述化合物10的结构通式为以下式(10):
[0045][0046]
其中,
[0047]
r1选自氢、羟基、卤素、c1
‑
c3烷基、c1
‑
c2烷基氧羰基c1
‑
c2烷基中的一种;
[0048]
r2选自氢、羟基、卤素、c1
‑
c3烷基、c1
‑
c2烷基氧羰基c1
‑
c2烷基中的一种;
[0049]
r3选自环己基甲酰基、环戊甲酰基、环庚烷甲酰基、1
‑
环己烯
‑1‑
碳酰基、3
‑
环己烯
‑1‑
碳酰基、四氢吡喃
‑4‑
甲酰基中的一种。
[0050]
进一步地,所述r1选自氢、羟基、甲基、乙基、丙基、乙酸甲酯基中的一种;所述r2选自氢、羟基、甲基、乙基、丙基、乙酸甲酯基中的一种;所述r3选自环己基甲酰基、环戊甲酰基中的一种。
[0051]
进一步地,在所述步骤一中,所述第一溶剂选自乙腈、二氯乙烷、四氢呋喃、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、乙酸钾、乙酸钠、n,n
‑
二异丙基乙胺中的至少一种;所述化合物1与所述化合物2、所述碱的摩尔比为1:0.8
‑
1.2:1.5
‑
3.5;所述反应时间为4
‑
8h。
[0052]
进一步地,在所述步骤二中,所述第二溶剂选自甲苯、二甲基亚砜、n,n
‑
二甲基甲酰胺、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸铯、碳酸钠中的至少一种;所述催化剂自pd2(dba)3或pd(oac)2;所述配体选自三环己基膦或1,3
‑
双(二环己膦基)丙烷;所述化合物3与1
‑
烯基己基硼酸、碱、催化剂、配体的摩尔比为1:1.1
‑
1.5:3
‑
5:0.05
‑
0.15:0.05
‑
0.15;所述反应时间为8
‑
15h。
[0053]
进一步地,在所述步骤三中,所述第三溶剂选自甲醇、乙醇、异丙醇、四氢呋喃、1,4
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、氢氧化钠、氢氧化钾中的至少一种;所述化合物4与碱的摩尔比为1:2.5
‑
4;所述反应时间为2
‑
4h。
[0054]
进一步地,在所述步骤四中,所述有第四溶剂为二氯甲烷、二氯乙烷、四氢呋喃、氯仿中的至少一种;所述氧化剂选自戴斯
‑
马丁氧化剂(dess
‑
martin)、重铬酸吡啶嗡盐、2
‑
碘酰基苯甲酸中的至少一种;所述化合物5与所述氧化剂的摩尔比为1:1.5
‑
3;所述反应时间为1
‑
2h。
[0055]
进一步地,在所述步骤五中,所述第五溶剂选自四氢呋喃、n,n
‑
二甲基甲酰胺、二甲基亚砜中的至少一种;所述碱选自正丁基锂、乙醇钠、叔丁醇钠、二甲基亚砜盐中的至少一种;所述化合物6与所述甲基三苯基溴化膦、所述碱的摩尔比为1:1.5
‑
2.5:1.5
‑
2.5;加入碱后,保温搅拌10
‑
50min;所述化合物6溶于所述第五溶剂的溶液加毕后,保温反应0.5
‑
1.5h。
[0056]
进一步地,在所述步骤六中,所述第六溶剂选自二氯乙烷、甲苯、二甲苯中的至少一种;所述催化剂为grubbs二代催化剂;所述化合物7与催化剂的摩尔比为1:0.15
‑
0.30;所
述第六溶剂与所述化合物7的比值为160
‑
320ml/g。
[0057]
进一步地,在所述步骤七中,所述第七溶剂选自甲醇、乙醇、叔丁醇、二氯甲烷、二氯乙烷中的至少一种;所述催化剂选自二氧化铂、钯碳中至少一种;所述化合物8与所述催化剂的摩尔比为1:0.1
‑
0.25。
[0058]
进一步地,在所述步骤八中,所述第八溶剂选自二氯甲烷、二氯乙烷、甲苯、n,n
‑
二甲基甲酰胺中的至少一种;所述酸液选自盐酸、三氟乙酸、硫酸中的至少一种;所述化合物9与所述酸液的摩尔比为1:8
‑
15。
[0059]
进一步地,在所述步骤九中,所述第九溶剂选自二氯甲烷、二氯乙烷、甲苯、二甲苯中的至少一种;所述碱选自碳酸钾、碳酸钠、三乙胺、n,n
‑
二异丙基乙胺中的至少一种;所述甲酰氯衍生物选自环己基甲酰氯、环戊甲酰氯、环庚烷甲酰氯、1
‑
环己烯
‑1‑
碳酰氯、3
‑
环己烯
‑1‑
碳酰氯、四氢吡喃
‑4‑
甲酰氯中的一种;所述化合物10与所述碱、所述甲酰氯衍生物的摩尔比1:4
‑
6:0.9
‑
1.2。
[0060]
本发明实施例具有如下优点:
[0061]
本发明实施例提供了一种苯并吡嗪类化合物及其合成方法,具体为苯环取代的1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类新化合物,可为苯并杂环类化合物药物的发展提供潜在的活性分子或活性分子砌块,从而进一步拓展该类活性药物的候选分子谱。本发明实施例针对现有技术中苯并杂环类化合物合成存在的问题,合成提供了一条操作简便,反应选择性高,所用试剂低毒安全的合成路线,为该类化合物的合成提供了可靠的参考,更具规模化扩大生产潜力。尤其构建七元杂环步骤,优选催化剂,降低了目标化合物的合成难度,减少了副反应,反应条件温和,操作简便,适用范围广,反应收率高,更具操作性。
具体实施方式
[0062]
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。除非另作定义,此处使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。
[0063]
本发明的目在之一在于提供一种苯环取代的1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类化合物,从而拓展苯并杂环类杀虫剂、杀菌剂的候选分子谱。
[0064]
下面通过具体实施例对本发明进行详细描述,但不限于此。
[0065]
本发明实施例公开了一种苯并吡嗪类化合物,具体为苯环取代的1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类新化合物,该新化合物的结构通式为以下式(11):
[0066][0067][0068]
其中,
[0069]
r1选自氢、羟基、卤素、c1
‑
c3烷基、c1
‑
c2烷基氧羰基c1
‑
c2烷基中的一种;
[0070]
r2选自氢、羟基、卤素、c1
‑
c3烷基、c1
‑
c2烷基氧羰基c1
‑
c2烷基中的一种;
[0071]
r3选自(环己基甲酰基)、(环戊甲酰基)、(环庚烷甲酰基)、(1
‑
环己烯
‑1‑
碳酰基)、(3
‑
环己烯
‑1‑
碳酰基)、(四氢吡喃
‑4‑
甲酰基)中的一种。
[0072]
优选地,所述r1选自氢、羟基、甲基、乙基、丙基、乙酸甲酯基中的一种;所述r2选自氢、羟基、甲基、乙基、丙基、乙酸甲酯基中的一种;所述r3选自环己基甲酰基、环戊甲酰基中的一种。
[0073]
随着苯并杂环类药物的长期使用,抗性、副作用等问题越来越凸显,苯并杂环类化合物活性分子谱需不断更新以应对药物发展的这一现状。由于1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类化合物的母环结构在许多疾病治疗中,作为活性基团起作用,在其母环结构上引入苯环,有望改变药物的空间结构以及脂溶性,从而改进其在生物体内的释放程度以及其与靶标的契合度,以获得高潜力的活性药物活性分子。
[0074]
本发明的目在之二在于提供一种苯环取代的1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类化合物的合成方法,该方法解决了现有技术中苯并杂环类化合物的合成所用试剂高毒高危,反应收率较低等问题,为该类化合物的制备提供一条反应条件温和、可操作性强、所用试剂低毒、反应收率高的合成路线。
[0075]
本发明实施例公开的上述一种苯并吡嗪类化合物的合成方法。具有式(11)作为结构通式的上述二苯甲基哌嗪类化合物的合成,采用以下合成路线:
[0076]
[0077]
上述一种苯并吡嗪类化合物的合成方法,具体地包括以下步骤:
[0078]
步骤一:将化合物1溶于第一溶剂中,加入化合物2及碱,体系回流反应至反应完全,反应结束,待体系温度冷至室温后过滤,旋干、纯化得到化合物3;
[0079]
步骤二:将所述化合物3溶于第二溶剂中,依次加入1
‑
烯基己基硼酸、碱和催化剂、配体,氮气保护下回流反应至反应完全,体系旋干、得到化合物4;
[0080]
步骤三:将所述化合物4溶于第三溶剂中,冰水浴下加入碱,加毕升至室温反应2
‑
4h,过滤后旋干溶剂,乙酸乙酯溶解,饱和食盐水洗,无水硫酸钠干燥,过滤,滤液旋干得到化合物5;
[0081]
步骤四:将所述化合物5溶于第四溶剂中,冰水浴下加入氧化剂,加毕后升至室温反应1
‑
2h;淬灭反应,水层用二氯甲烷萃取,合并有机层,饱和食盐水洗涤,干燥,过滤,滤液浓缩得到化合物6;
[0082]
步骤五:将甲基三苯基溴化膦溶于第五溶剂中,
‑
5~0℃下滴入碱,滴毕后保温搅拌,而后加入所述化合物6溶于所述第五溶剂的溶液,保温反应至反应完全,用饱和氯化铵淬灭反应,水层用乙酸乙酯萃取,合并有机层用饱和食盐水洗,无水硫酸钠干燥,过滤,滤液浓缩,硅胶柱层析得到化合物7;
[0083]
步骤六:将所述化合物7溶于第六溶剂中,加入催化剂,回流反应过夜至反应完全,减压旋干溶剂,硅胶柱纯化得到化合物8;
[0084]
步骤七:将所述化合物8溶于第七溶剂中,加入催化剂,室温氢化反应至完全;过滤,甲醇洗滤渣,滤液旋干溶剂后,硅胶柱纯化得到化合物9;
[0085]
步骤八:将所述化合物9溶于第八溶剂中,冰水浴下加入酸液,滴毕后撤去冰浴,室温反应至反应完全;慢慢加入饱和碳酸氢钠溶液淬灭反应后,二氯甲烷萃取后饱和食盐水洗,无水硫酸钠干燥;过滤,减压蒸去溶剂后得到化合物10,未经纯化,直接用于下步;
[0086]
步骤九:将上步所得化合物10溶于第九溶剂中,冰水浴下依次加入碱和甲酰氯衍生物,室温反应过夜至完全,后加入饱和碳酸氢钠溶液淬灭反应;水层用二氯甲烷萃取,合并有机层,无水硫酸钠干燥;旋干溶剂后,硅胶柱纯化得到目标化合物11。
[0087]
1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂衍生物的合成关键在于七元环的构建,现有技术中,采用先构建氮杂七元环,后构建哌嗪环的合成策略合成该类化合物,在七元环的构造中,用到剧毒且具高爆炸性的叠氮钠试剂,不仅安全隐患高,操作上极具繁琐性,合成收率也较低,不利于工业化扩大生产。这在一定程度上限制了1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类化合物的推广应用。就现有技术的这一现状,本发明基于1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类化合物的母环框架,进行合成策略的调整,先构建二苯甲基哌嗪中间体,再构建七元环框架,采用1
‑
溴
‑2‑
(溴(苯基)甲基)苯与保护基团保护的哌嗪作为原料,进行反应,反应在碱的存在下进行亲核取代,得到二苯甲基哌嗪中间体。对于七元环的构造,现有技术中含哌嗪环的底物构造七元环合成类似物时,常采用多聚磷酸高温合环,如4
‑
苄基
‑6‑
羟基
‑1‑
(3
‑
苯基丙基)哌嗪
‑2‑
酮在多聚磷酸存在下,180℃高温反应,该条件下部分底物易碳化,使得副反应较多,反应较杂,反应收率仅32%。本发明实施例通过官能团的转换形成含两个烯键的中间体,而后在grubbs催化剂作用下,发生复分解反应,失去一分子烯烃,同时形成新的分子内烯烃,实现合环。该过程所用催化剂价廉且易得,反应选择性高,操作简便,对底物基团的容忍度高,可
以为多种1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂衍生物的合成提供参考。历经烯烃还原、脱保护步骤合成得到吡嗪并[1,2
‑
b][2]苯并氮杂框架后,在碱存在下,与相应甲基酰氯衍生物反应,即可得到目标化合物。总的来说,整条合成路线操作简便,反应活性高,反应具有专一性,所用原料均市售可得,未用到高毒试剂,具有工业扩大生产的潜力。
[0088]
在上述步骤一中,所述第一溶剂选自乙腈、二氯乙烷、四氢呋喃、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、乙酸钾、乙酸钠、n,n
‑
二异丙基乙胺中的至少一种;所述化合物1与所述化合物2、所述碱的摩尔比为1:0.8
‑
1.2:1.5
‑
3.5;所述反应时间为4
‑
8h。
[0089]
在上述步骤二中,所述第二溶剂选自甲苯、二甲基亚砜、n,n
‑
二甲基甲酰胺、1,2
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸铯、碳酸钠中的至少一种;所述催化剂自pd2(dba)3或pd(oac)2;所述配体选自三环己基膦或1,3
‑
双(二环己膦基)丙烷,并进一步优选1,3
‑
双(二环己膦基)丙烷;所述化合物3与1
‑
烯基己基硼酸、碱、催化剂、配体的摩尔比为1:1.1
‑
1.5:3
‑
5:0.05
‑
0.15:0.05
‑
0.15;所述反应时间为8
‑
15h。
[0090]
在上述步骤三中,所述第三溶剂选自甲醇、乙醇、异丙醇、四氢呋喃、1,4
‑
二氧六环中的至少一种;所述碱选自碳酸钾、碳酸钠、氢氧化钠、氢氧化钾中的至少一种;所述化合物4与碱的摩尔比为1:2.5
‑
4;所述反应时间为2
‑
4h,进一步优选2h。
[0091]
在上述步骤四中,所述有第四溶剂为二氯甲烷、二氯乙烷、四氢呋喃、氯仿中的至少一种;所述氧化剂选自戴斯
‑
马丁氧化剂(dess
‑
martin)、重铬酸吡啶嗡盐中的至少一种,并进一步优选dess
‑
martin作为氧化试剂;所述化合物5与所述氧化剂的摩尔比为1:1.5
‑
3;所述反应时间为1
‑
2h,并进一步优选为1h。
[0092]
在上述步骤五中,所述第五溶剂选自四氢呋喃、n,n
‑
二甲基甲酰胺、二甲基亚砜中的至少一种;所述碱选自正丁基锂、乙醇钠、叔丁醇钠、二甲基亚砜盐中的至少一种;所述化合物6与所述甲基三苯基溴化膦、所述碱的摩尔比为1:1.5
‑
2.5:1.5
‑
2.5;加入碱后,保温搅拌10
‑
50min,优选20min;所述化合物6溶于所述第五溶剂的溶液加毕后,保温反应0.5
‑
1.5h,优选0.5h。
[0093]
在上述步骤六中,所述第六溶剂选自二氯乙烷、甲苯、二甲苯中的至少一种;所述催化剂为grubbs二代催化剂;所述化合物7与催化剂的摩尔比为1:0.15
‑
0.30;所述第六溶剂与所述化合物7的比值为160
‑
320ml/g。
[0094]
在上述步骤七中,所述第七溶剂选自甲醇、乙醇、叔丁醇、二氯甲烷、二氯乙烷中的至少一种;所述催化剂选自二氧化铂、钯碳中至少一种;所述化合物8与所述催化剂的摩尔比为1:0.1
‑
0.25。
[0095]
在上述步骤八中,所述第八溶剂选自二氯甲烷、二氯乙烷、甲苯、n,n
‑
二甲基甲酰胺中的至少一种;所述酸液选自盐酸、三氟乙酸、硫酸中的至少一种;所述化合物9与所述酸液的摩尔比为1:8
‑
15。
[0096]
在上述步骤九中,所述第九溶剂选自二氯甲烷、二氯乙烷、甲苯、二甲苯中的至少一种;所述碱选自碳酸钾、碳酸钠、三乙胺、n,n
‑
二异丙基乙胺中的至少一种;所述甲酰氯衍生物选自环己基甲酰氯、环戊甲酰氯、环庚烷甲酰氯、1
‑
环己烯
‑1‑
碳酰氯、3
‑
环己烯
‑1‑
碳酰氯、四氢吡喃
‑4‑
甲酰氯中的一种;所述化合物10与所述碱、所述甲酰氯衍生物的摩尔比1:4
‑
6:0.9
‑
1.2。
[0097]
由以上技术方案可知,本发明提出了一种苯环取代的1,2,3,4,6,11,12,12a
‑
八氢苯并[e]吡嗪并[1,2
‑
a]氮杂类新化合物,可为苯并杂环类化合物药物的发展提供潜在的活性分子或活性分子砌块,从而进一步拓展该类活性药物的候选分子谱。本发明针对现有技术中苯并杂环类化合物合成存在的问题,为该化合物的合成提供了一条操作简便,反应选择性高,所用试剂低毒安全的合成路线,为该类化合物的合成提供了可靠的参考,更具规模化扩大生产潜力。尤其构建七元杂环步骤,优选催化剂,降低了目标化合物的合成难度,减少了副反应,反应条件温和,操作简便,适用范围广,反应收率高,更具操作性。
[0098]
实施例1
[0099]
以下,以r1选自氢;r2选自氢;r3选自(环己基甲酰基);为例,说明本发明实施例中的上述一种苯并吡嗪类化合物的合成方法。
[0100]
步骤一:化合物3的合成
[0101][0102]
将化合物1(20g,61.34mmol,1eq)溶于乙腈(150ml),加入化合物2(15.85g,61.36mmol,1eq)及碳酸钾(17g,123.0mmol,2eq),体系回流反应4小时。反应结束,待体系温度冷至室温后过滤,旋干溶剂后硅胶柱层析得到白色固体27.51g,即为化合物3,收率87.4%,纯度98.1%。
[0103]
化合物3的结构表征数据如下:
[0104]
[m+h]
+
=503.16
[0105]1h
‑
nmr(300mhz,cdcl3)δ7.79(m,1h),7.48(m,3h),7.28(m,3h),7.20(m,1h),7.06(m,1h),5.42(m,1h),4.28(m,2h),3.95(m,2h),3.08(m,3h),2.61(m,2h),2.00(m,3h),1.43(s,9h)。
[0106]
步骤二:化合物4的合成
[0107][0108]
将化合物3(27.51g,53.63mmol,1eq)溶于甲苯(300ml),依次加入1
‑
烯基己基硼酸(8.24g,64.39mmol,1.2eq)、碳酸钾(22.24g,160.91mmol,3eq)、pd2(dba)3(4.91g,5.365mmol,0.1eq)和1,3
‑
双(二环己膦基)丙烷(2.34g,5.365mmol,0.1eq),体系氮气置换三次。氮气保护下回流反应过夜。次日,体系旋干溶剂后硅胶柱层析得到淡黄色液体26.09g,即为化合物4,收率94.00%,纯度97.9%。
[0109]
化合物3的结构表征数据如下:
[0110]
[m+h]
+
=507.32
[0111]1h
‑
nmr(300mhz,cdcl3)δ7.73(q,1h),7.38(d,j=5.7hz,2h),7.13
‑
7.35(m,6h),6.80(m,1h),5.91(m,1h),5.20(m,1h),4.30(m,2h),3.95(m,1h),3.0(m,2h),2.57(m,2h),2.22(q,2h),1.97(m,3h),1.49(q,2h),1.43(s,9h),1.38(m,4),0.98(dd,j=7.8hz,3h)。
[0112]
步骤三:化合物5的合成
[0113][0114]
将化合物4(4.78g,9.43mmol,1eq)溶于甲醇(50ml)中,冰水浴下加入碳酸钾(3.91g,28.29mmol,3eq),加毕后撤去冰水浴室温反应2小时。过滤后旋干溶剂,60ml乙酸乙酯溶解,饱和食盐水洗,无水硫酸钠干燥。过滤,减压蒸去溶剂后得到4.45g淡黄色液体,即为化合物5,收率为93.34%,纯度为92.00%。
[0115]
步骤四:化合物6的合成
[0116][0117]
将上步所得化合物5的淡黄色液体(4.09g,8.10mmol)溶于二氯甲烷(40ml),冰水浴下分三次加dess
‑
martin氧化剂(8.6g,24.90mmol,2.5eq),加毕后撤去冰水浴室温反应1h。加入饱和碳酸氢钠和硫代硫酸钠的混合溶液淬灭反应后,水层用二氯甲烷萃取,合并有机层后用饱和食盐水洗,无水硫酸钠干燥。过滤,减压蒸去溶剂后,得到3.68g淡黄色液体,即为化合物6,收率为89.78%,纯度为91.30%。
[0118]
步骤五:化合物7的合成
[0119][0120]
将甲基三苯磷溴(5.19g,14.54mmol,2eq)溶于四氢呋喃(120ml),0℃下慢慢滴入正丁基锂(5.80ml,14.54mmol,2eq),滴毕后0℃搅拌20min,后慢慢滴入上述得到的化合物6(3.68g,7.27mmol,1eq)的四氢呋喃溶液(20ml),保温反应0.5h后,用饱和氯化铵淬灭反应。水层用乙酸乙酯萃取,合并有机层后用饱和食盐水洗,无水硫酸钠干燥。过滤,减压蒸去溶剂后,硅胶柱层析得到2.61g淡黄色液体,即为化合物7,收率为76.37%,纯度为98.12%。
[0121]
化合物7的结构表征数据如下:
[0122]
[m+h]
+
=461.32
[0123]1h
‑
nmr(300mhz,cdcl3)δ7.63(m,1h),7.12
‑
7.34(m,9h),6.77(t,j=30.3,1h),6.39(m,2h),5.58(m,2h),4.88(s,1h),3.80(m,2h),3.32(m,3h),3.02(brs,1h),2.75(m,2h),1.40(s,9h),1.38(m,4),0.98(m,3h)。
[0124]
步骤六:化合物8的合成
[0125][0126]
将化合物7(9.38g,19.97mmol,1eq)溶于甲苯(2000ml),加入grubbs二代催化剂(3.3g,3.89mmol,0.19eq),回流反应过夜。减压旋干溶剂,硅胶柱层析得到白色固体5.20g,收率68.00%,纯度98.33%。
[0127]
化合物8的结构表征数据如下:
[0128]
[m+h]
+
=379.24
[0129]1h
‑
nmr(300mhz,cdcl3)δ7.12
‑
7.34(m,9h),6.39(m,1h),5.58(m,1h),4.88(s,1h),3.80(m,2h),3.32(m,2h),3.02(brs,1h),2.75(m,2h),1.40(s,9h)。
[0130]
步骤七:化合物9的合成
[0131][0132]
化合物8(3.1g,7.97mmol,1eq)溶于甲醇(30ml),加入三水合二氧化铂(0.3g,1.32mmol,0.16eq)后,氢气置换体系三次,而后氢气氛中,室温下进行氢化反应。3h后反应毕,过滤,甲醇洗滤渣,旋干滤液,硅胶柱层析得到2.23g白色固体化合物,即为化合物9,收率72.40%,纯度98.03%。
[0133]
步骤八:化合物10的合成
[0134][0135]
将化合物9(3.06g,7.93mmol,1eq)溶于二氯甲烷(30ml),冰水浴下慢慢加入三氟乙酸(6ml,79.3mmol,10eq),滴毕后撤去冰浴室温反应2h。慢慢加入饱和碳酸氢钠溶液淬灭反应后,二氯甲烷萃取后饱和食盐水洗,无水硫酸钠干燥。过滤,减压蒸去溶剂后得到淡黄色固体,即为化合物10,未经纯化直接投下步。
[0136]
步骤九:目标化合物11(r1=h,r2=h,)的合成
[0137][0138]
上一步化合物10溶于二氯甲烷(30ml),冰水浴下依次加入三乙胺(5.5ml,39.65mmol,5eq)和环己基甲酰氯(1.16,7.93mmol,1.0eq),室温反应过夜后加入饱和碳酸氢钠溶液淬灭反应。水层用二氯甲烷萃取,合并有机层后无水硫酸钠干燥。旋干溶剂后硅胶柱层析得到2.39g白色固体,即为上述化合物11,收率76.52%,纯度98.66%。
[0139]
化合物11的结构表征数据如下:
[0140]
[m+h]
+
=389.26
[0141]1h
‑
nmr(300mhz,cdcl3)δ7.29
‑
7.43(m,5h),7.03
‑
7.15(m,3h),6.77(d,j=7.2hz,1h),5.53(s,1h),4.30(m,1h),3.58
‑
3.66(m,1h),3.18
‑
3.42(m,4h),3.02(m,2h),2.82(m,
1h),2.39(m,3h),1.20
‑
1.98(m,10h)。
[0142]
实施例2
‑
11及对比例1
‑5[0143]
实施例2
‑
7、对比例1
‑
4的步骤与上述实施例1相同,仅改变了步骤二的部分工艺条件。为使结果更加直观明了,实施例2
‑
7、对比例1
‑
4采用了列表的方式,对本发明进行了具体说明。
[0144]
表1反应条件对中间体化合物8(r1=h,r2=h)收率的影响
[0145][0146]
该步反应的关键在于grubbs二代催化剂的用量,如实施例1
‑
3所示,当其用量为0.15
‑
0.3eq时反应收率较为理想;当其用量降低至对比例1中的0.08eq,反应收率明显降低。该反应可在多种中高沸点溶剂中实现,如实施例4
‑
5所示,当其采用对比例2中的低沸点溶剂进行时,反应速率大幅度降低,同等反应时间下的反应收率下降;在该反应中,grubbs二代催化剂可以有效的提高反应活性,当其采用对比例3中的grubbs一代催化剂进行反应,反应活性的提高并不明显,反应收率降低;在该反应中,溶剂量也较为关键,当其溶剂量为160ml/g至320ml/g时,反应结果较为理想,如实施例6
‑
7所示,当溶剂量降低对比例4中的86ml/g时,反应收率降低明显。
[0147]
虽然,上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1